Prepared for Bottlenecks 2021 in Denver CO, reposted here.
Summary
Nanoscale science and technology broadly has made considerable progress on areas ranging from mRNA vaccine delivery that are impacting our lives, to carbon nanotube computers that I think soon will. But the idea of general purpose nano-manufacturing as a kind of stand-alone technology has fallen out of favor. The purpose of this document is to understand why that happened, and what kinds of actions by organizers and funders of R&D might reboot the field of general purpose nanomanufacturing.
We’ll conclude, at the highest level:
- The nanotechnology field has largely forgotten or disavowed one of its original motivating ambitions — positional chemistry — at least at the level of explicitly articulated directions, if not the tacit motivations of individual researchers
- Progress has been made, however, on enabling technical factors for those ambitions, due to serendipitous developments in the meantime, particularly DNA origami
- Going further will require a “systems” approach that the field is not fully embracing yet
- Identifying and crystallizing around a set of ARPA-like systems engineering projects could help jumpstart progress, and there are several viable candidates for such projects
- Discovering commercial killer apps for the most general, programmable nano-fabrication methods could also jumpstart progress on the systems challenges, but to get this we’ll likely need to start with a pre-commercial “foundry” and design enablement model, to help discover what those killer apps could be in the first place
To preview where we’ll go in a bit more detail, by the end of this essay we’ll have concluded the following:
- There is a strong theoretical case for at least some forms of positional chemistry being possible
- Actually in two different initial categories
- Vacuum mechanosynthesis
- This is being tried secretively in Canada by enthusiasts
- Ribosome-like molecular additive manufacturing
- This is not being seriously tried yet by any well-supported research program
- Vacuum mechanosynthesis
- With decades of focused, coordinated work informed by systems goals, either of these could potentially lead to forms of positional chemistry
- It is not yet clear exactly how general-purpose these could be
- Actually in two different initial categories
- Early attempted refutations (Smalley debate) raised straw man objections; they don’t rule out either path
- However, they created a taboo against working on positional chemistry or funding it
- Because positional chemistry is also a very hard systems problem, beyond the reach of any one scientist or lab, individual courageous researchers can’t escape this sociological trap by quietly showing a demo
- This means that you don’t see many people even submitting grant proposals or theoretical papers on positional chemistry, let alone making major experimental progress on it
- In such a situation, one needs a DARPA-like coordinated series of programs to unlock progress
- Progress on a “ribosome-like” mode of positional chemistry (“molecular 3D printing”) is enabled by recent advances like DNA origami, and more modular forms of protein engineering
- The field needs more crystalline goals and subgoals, but early developments are likely NOT good as commercial ventures
To be clear, I’m not saying positional chemistry is necessarily a “be all and end all” technology. I’m just asking if we can pinpoint why it may have become stalled as a research field, and what it might take to jump start it as a research field. This then might, or might not, lead to big real world applications, many decades down the line. It’s research!
Update on items found since 2021:
This old startup
http://molecubotics.com/tech-docs/dgap.html
http://molecubotics.com/tech-docs/asm-roadmap-0.7.html
http://www.n-a-n-o.com/nano/cda-news/cda-news.html
https://web.archive.org/web/20210510210416/http://molecubotics.com/
This shows the EA community starting to grapple with the area (again)
Two papers that are explicitly in related directions
https://www.science.org/doi/10.1126/science.abm1183
https://www.science.org/doi/abs/10.1126/scirobotics.abn5459
The positional chemistry concept and its history
Here is a snippet from the great physicist Richard Feynman’s 1959 talk “There’s Plenty of Room at the Bottom”:

This immediately raises a few key points that easily get lost. First, the novelty of the size scale “nano”, or 1e-9 meters, per se, was not the main thrust of Feynman’s interest. His main point was arguably a notion of programmability, design control or directness of construction at the atomic and molecular level.
The reason “nano” comes up at all is because it’s a size scale a few atoms across — chemical bonds between atoms in a molecule are about 1/10th of a nanometer (nm) long. Here are some nice illustrations of how big 1 nm is in the context of various biomolecules (be sure to check the scale bars in each image):
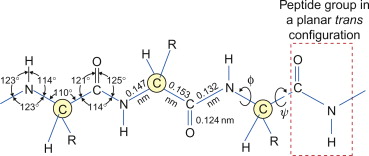
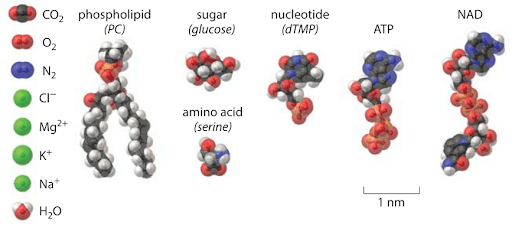
Feynman was asking: rather than going through the usual bespoke, complex and “mysterious” recipes of the chemist (“mixing and shaking” vials of liquid) to synthesize molecules, what if we could have a kind of direct write approach to arranging atoms and molecules? It was a very “printer”-like or assembly-line-like concept: “put the atoms down where the chemist says”, and “maneuver” things atom by atom. To do that in full generality would require positioning and orienting molecules, and moving them around in a directed way, with a precision of perhaps a fraction of a nanometer.
So to picture this conceptually, with molecules represented as legos, Feynman thinks of standard chemistry as something like this — you mix a lot of molecules together and, by bumping randomly into each other as they jiggle around in solution, they can form certain products. But you can only control the outcome by relying on their intrinsic shapes, and by deciding which collections you mix in which order and under which overall conditions

As we’ll see, you can go far with this approach, not only in synthetic chemistry to form complex covalently bonded molecules, but also in “supra-molecular” chemistry, particularly when using information-rich “bricks” like DNA strands, which can self-assemble into intricate higher-order structures.
Whereas Feynman’s articulation of nanotechnology would rely on direct positioning and mechanically driven assembly of molecules, conceptually more like this

Perhaps not incidentally, the first computer numerically controlled (CNC) milling machines had been developed in the decade leading up to 1959

Feynman also pointed out both a theoretical limitation, “of course you can’t put them so they are chemically unstable”, and, if you will, a market limitation: “by the time I get my devices working… [the chemist] will have figured out how to synthesize absolutely anything, so that this will really be useless”. In other words, it was not yet clear what the unique value proposition of direct write construction of matter at the atomic scale would be, relative to, say, the more traditional methods of chemistry or biochemistry, and when it would be possible to realize that value proposition. This is still a problem today, as we’ll see, for bootstrapping the field.
(Feynman also offered a specific notion of how this could be made to work, namely a teleoperated machine shop that would construct and operate a smaller machine shop that would in turn construct a smaller machine shop and so on, MEMS style. There is at least one extant proposal by JS Hall, also covered in his book “Where is my flying car”, to actually do it that way. Probably there are much handier ways than that now available, though.)
That was 1959, only a few years after the discovery of the double helical structure of DNA, and a few years before the discovery of the genetic code and the “central dogma” of molecular biology. In the central dogma of molecular biology, DNA chains codes for RNA chains, which in turn direct molecular machines called ribosomes
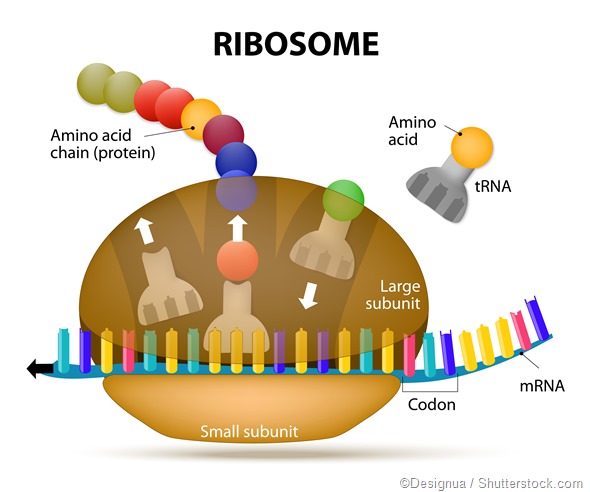
to construct more chemically complex chains called proteins, which fold into atomically precise machines that are the workhorses of the cell
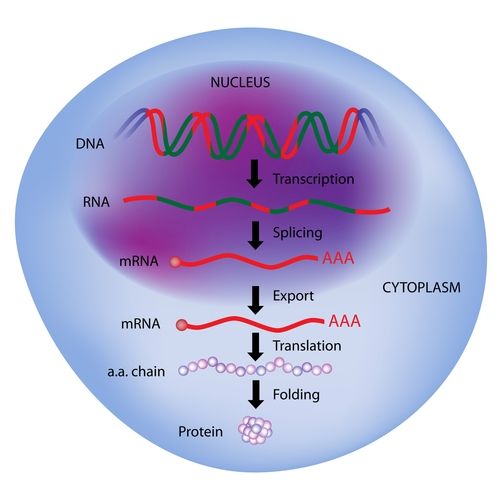
The following few decades would thus see a totally unprecedented explosion of basic knowledge of the biomolecular machines in cells and how they work.
So, twenty years later, nanotechnology’s next key visionary, Eric Drexer (who had been trained in aerospace engineering, but had spent many of his days in the library reading about the past two decades of revolution in biochemistry) wrote a paper “Molecular engineering: An approach to the development of general capabilities for molecular manipulation” proposing to apply engineering approaches to the substrate of biochemistry. In that paper, he included the following table, crystallizing a pattern of thinking implicit in the reams of biochemical literature he was reading: that we can think of biological macromolecules, like the ribosome, as a kind of “molecular machine”, one that directs chemistry by using molecular geometries and mechanical motion thereof to precisely control which chemical reactions happen, where, and when

A great example of this analogy is the bacterial flagellar motor
and another is the bacterial pilus
[Drexler was not the only researcher to consider abstract generalizations of the ribosome. In his paper on Constructor Theory, for example, David Deutch includes this passage

[I don’t think polyketide synthases were yet known, but they would have provided another good example, something like an “assembly line”, as engineers have since realized.]
Anyway, this identification of biological “molecular machines” raised the question of whether engineers can utilize these same basic principles and primitives, like nucleic acid and amino acid chains, in a more designable or engineerable way, making the analogies between chemical/biological machines and mechanical machines even more direct via design. Drexler speculated that although predicting the folds of natural proteins from their sequences was hard (borne out by it taking until 2020 for DeepMind’s to have staked a claim to solving this problem, at least small proteins similar to those in existing biological families), potentially, by deliberately restricting the sequences and design rules, engineers could devise a more constrained subspace of proteins whose folds would be designable in a more facile way. In other words, engineering design could be easier than general prediction. This is similar to how we got airplanes to work long before we fully understood the aerodynamics and control strategies of natural bird or insect flight, nor could simulate a bird or insect.
Drexler continued applying the strategic framework of engineering (modularity, design constraints, functional requirements on components, basically a “systems engineering” mindset) to the issue of programmable biomolecular construction, and by the early 1990s had published a technical book, called Nanosystems. Nanosystems mostly covered the physics of what he saw as an illustrative example of the kind of long-term design space that would embody “Feynman’s vision” of direct-write atomically precise construction and manipulation. His strategy in that and his other books seemed to be to try to lay out a sufficiently compelling long-term vision, establishing the existence of a desirable end-point, assuming that the government would then fund and organize all the work necessary to get there. The book was embraced by the engineering-minded, winning an award for best computer science book of 1992, but many mainstream chemists and biologists seemed to find it rather divorced from the practical experimental realities of the time. Notably, only a few pages of the book are devoted to how we can get from here to there, starting with today’s tools.
Meanwhile, his 1986 popular book Engines of Creation laid out a larger picture proposing broad, transformational implications of a direct write approach to constructing molecular matter, introducing the notion of molecular assemblers as molecular machines that could build other molecular machines and structures by mechanically guiding chemical reactions. Could we reduce physical construction to something more like software? Could factories with nanoscale machinery and molecularly precise components massively increase the throughput and capabilities of manufacturing across many fields (there are some principled reasons to think that they could, e.g., around the idea of advantages in the speed of “exponential manufacturing” when using nanoscale systems versus macroscale)?
Here are some quotes. He starts by saying that by using amino acid chains (proteins) as a design and engineering substrate, rather than just studying natural proteins, we could start to engineer nanoscale machines, following the narrative of his 1979 paper. He then asks whether such protein engineering could be bootstrapped to construct mechanical machines that can synthesize other molecules programmably based on outside instructions, via a sequence of directed mechanical motions, much as ribosomes synthesize protein chains under the instruction of an RNA chain. He then goes on to suggest that such protein machines could construct machines made out of other chemical building blocks, which in turn could be even more powerful as programmable constructors, using increasingly designable and understandable mechanical components and operations


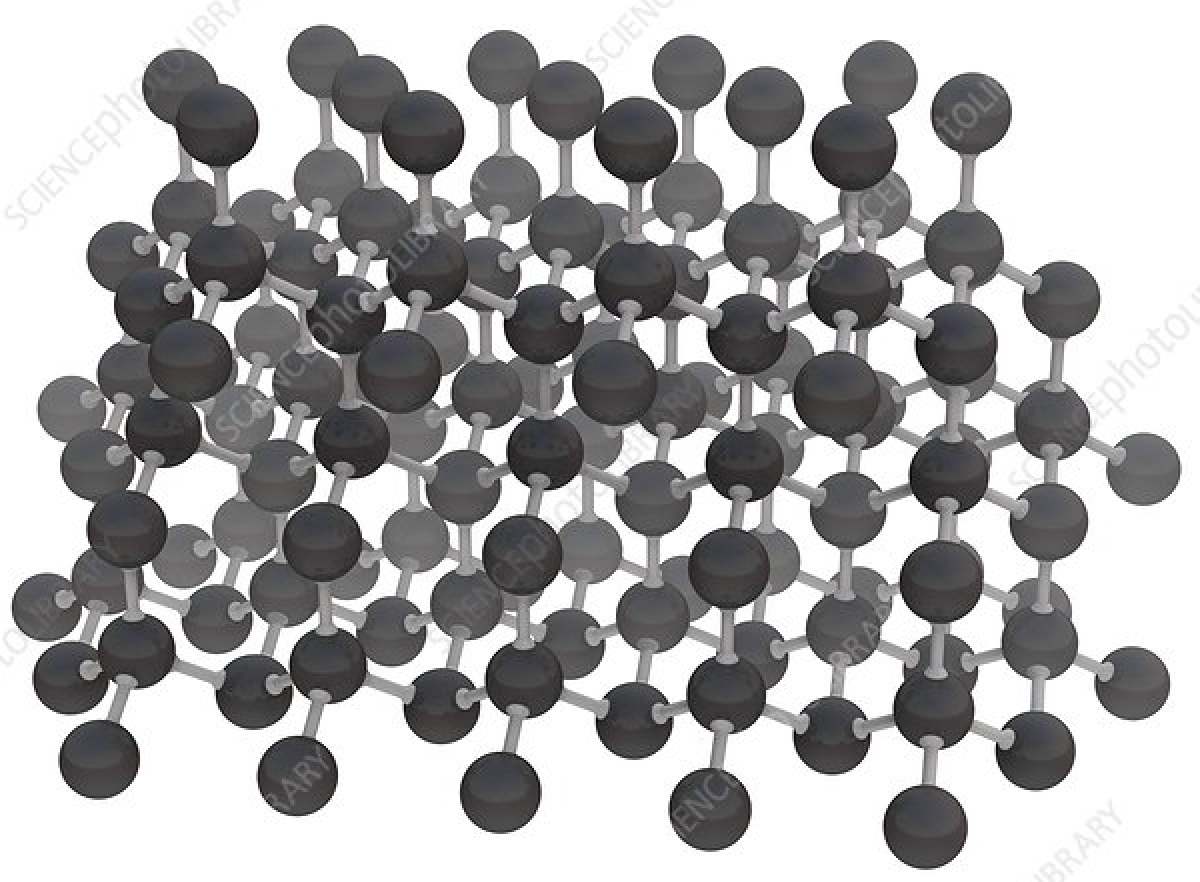
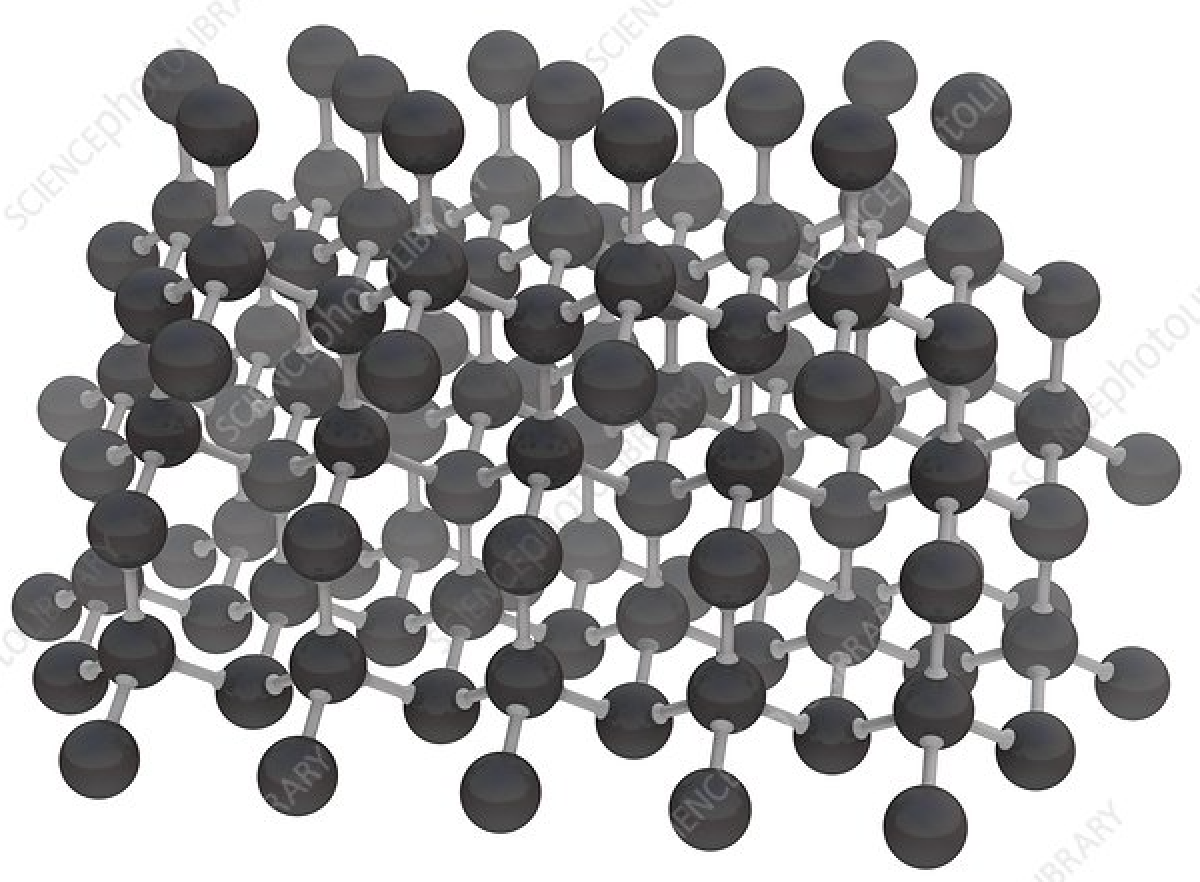
Note how he specifically suggests that such machines could construct tightly bonded, rigid assemblies of pure carbon, i.e., bootstrapping from floppy biological molecules to a pristine and rigid mechanical machine world. Here is the molecular structure of diamond, each sphere being a carbon atom
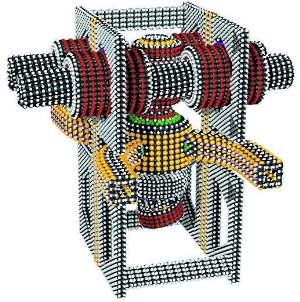
and here is a concept of the kind of machine (an “acetylene sorting pump”, supposedly) that one could construct if one could add and remove carbon and other atoms from such 3D lattices site by site under direct positional control
Could these in turn be used to create fleets of nanoscopic assembly lines that could themselves create macroscopic structures and materials? This kind of forward-looking thinking started to get people quite excited. Hence Neil Stephenson’s wonderful sci-fi novel The Diamond Age, published 1995, in which such technology had been used to effectively end physical scarcity and realize the notion of a “matter compiler” as well as insanely powerful AI on small but wildly powerful and energy efficient tablet computers.
At the same time, the prospect of direct write programmable atomic scale fabrication was actually looking increasingly reasonable, at least in the earliest two-dimensional demos, as scanning probe microscopy systems — see Michael Nielsen’s great piece on these — were being used to move individual atoms around on surfaces to make arbitrary user-defined patterns, as in the famous work of Don Eigler from IBM circa 1989. The little bumps here are individual atoms imaged and mechanically manipulated one by one with a scanning tunneling microscope to form these patterns on a surface


By 2000, President Bill Clinton was excited by the prospects, and launched the National Nanotechnology Initiative (NNI) in a speech at Caltech:

(Note the presence of Richard Symonds in the transcript, presumably the celebrity exercise instructor, rather than physicist Richard Feynman.)
This led to billions of dollars of investment in the field, proliferating the number of academic researchers working on nanoscale science and technology, and giving them more widespread and democratized access to experimental infrastructure and tools like scanning probe microscopes. The NNI has undoubtedly contributed to many advances, from lipid nanoparticles for mRNA delivery, to quantum dot fluorescent labels for bio-imaging and nanoparticle solar cells, advances towards carbon nanotube and graphene based 3D low energy beyond-silicon computing, understanding of materials properties like superconductivity, the burgeoning field quantum information/computing, and so on. You can get a sense of the range of nanoscale science and engineering it supports in its 2020 budget supplement doc.
In 2011, Mihail Roco, one of the architects of the NNI, included in a publication (now cited over 600 times) the following figure, showing a prospective “four generations” of nanotechnology. In the 3rd generation, slated for the 2010-2015 period, “guided assembling” was still highlighted prominently, as was the construction of “hierarchical architectures” that would cross from the nanoscale up to the level of complex integrated devices, as was doing so at the “atomic design” level in the 2015-2020 period, leading to “large complex systems from nanoscale”:
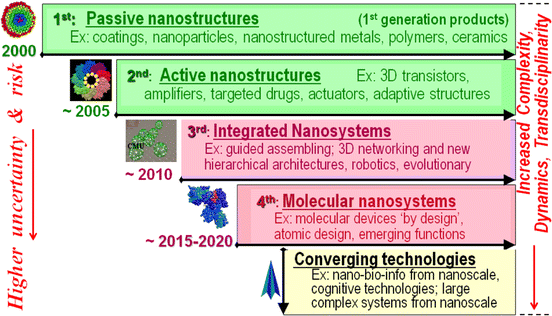
That’s an ambitious vision. “Guided assembling” sounds a lot like “positional assembly”, “large complex systems from nanoscale” sounds a lot like scalable atomically precise manufacturing of large complex objects, and “atomic design” sounds a lot like Feynman’s original talk. At the same time, this doesn’t depict a concrete technical roadmap for doing these things, nor precise definitions of these terms (which can be justified from a “let all flowers bloom” approach on the part of a funder).
Flashing forward to 2021, it is at best not clear that much of this ambitious “nano-systems” agenda is panning out, and in a recent proposal to add a technology directorate to the NSF, “nanotechnology” is not mentioned as a priority area, even as biotechnology, advanced manufacturing, materials and quantum information are given central importance, as is semiconductor technology (computer chips).
What is nanotechnology actually supposed to accomplish?
Given this last point, one might ask what nanotechnology is actually supposed to add over and above biotechnology or advanced manufacturing and so on. I don’t want this to be the main point here, as I want to focus on specific technical aspects of some basic enabling steps or principles, rather than hyperventilating about the grand long-term vision— indeed, I’m not even sure what I think about the grand long-term vision. But it is worth at least mentioning a possibility for the long-term trajectory.
To try to answer this, let me start by borrowing a bit from the 2007 Battelle Roadmap on atomically precise productive nanosystems, which we’ll return to later. Notably, it defines a few key criteria for what should count as uniquely within its proposed direction.
Atomically precise structures: “structures that consist of a specific arrangement of atoms” (as opposed to most, say, nanoparticles which are defined by an overall shape and size but don’t specify the position and identity of every individual atom via its covalent bonding pattern)
Atomically precise productive nanosystems: “functional nanosystems (interacting nanoscale structures, components, and devices that process material, energy, or information) that make atomically precise structures, components, and devices under programmable control, that is, they are advanced functional nanosystems that perform atomically precise manufacturing”
Defining positional chemistry
While we’re defining things, let’s also be a bit specific in defining what we actually mean here by the term “positional chemistry”. I define it to require:
the ability to choose between forming otherwise chemically equivalent covalent bonds, under flexible, programmable mechanical control — at least for covalent bonds BETWEEN certain finite sized “building blocks” that combine covalently to form the target object (and ideally for many of the bonds in the entire target object)
David Leigh quoted similarly in a recent talk, building on a sentence in Drexler’s 1981 PNAS paper, “machine-like operation… to select individual bonds on the basis of position alone”… “Further, where chemists must resort to complex strategies to make or break specific bonds in large molecules, molecular machines can select individual bonds on the basis of position alone. Conventional organic chemistry can synthesize not only one-, two-, and three-dimensional covalent structures but also exotic strained and fused rings. With the addition of controlled site-specific synthetic reactions, a broad range of large complex structures can doubtless be built.”
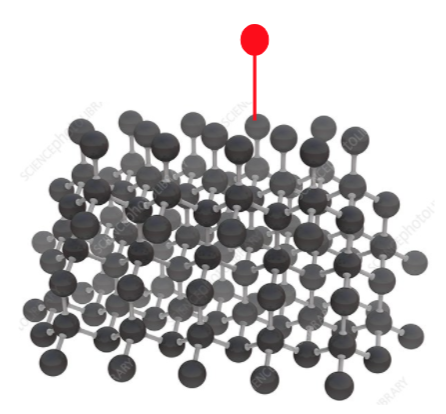
So to be clear, consider very conceptually this diamond structure
We’d like to be able to add a bond to an added chemical (in red) here
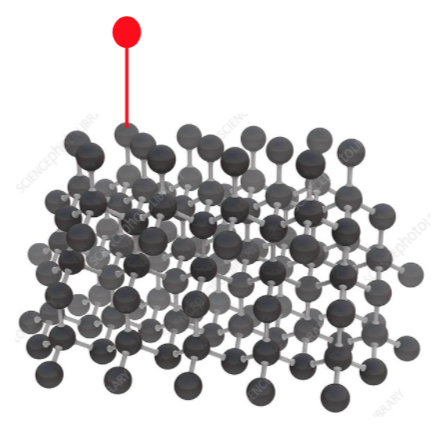
instead of the otherwise chemically equivalent site here
Now in the case of the ribosome analogy, it only controls a certain subset bonds, not any old bond in the structure, namely the ones needed to determine the linear chain of amino acid building blocks in the protein, namely the bonds between them. It does not get to arbitrarily choose the bonds that exist inside the individual amino acids. That’s still a form of positional chemistry in my mind, even if not as powerful as being able to choose every single bond.
Now let’s bump back up to the broader question of “what is nanotechnology supposed to accomplish”? What the broader vision originally associated with positional chemistry looks for is the ability to build complex, atomically precise machines and systems, which themselves can be programmed to build other similarly complex, atomically precise machines and systems. What this leads to, then, is a notion of a fairly general purpose, programmable, atomically precise form of manufacturing.
Going further, it could be a form of manufacturing that is capable of some kind of exponential scaleup. In particular, one could be looking for nanoscale machinery that can be programmed to produce machinery as complex as itself, or indeed a copy of itself — like a factory for making factories.
The “atomically precise” part means that this manufacturing system can exert a kind of ultimate level of chemical control, making machines that are as precise as any that could be made. The “productive” part means that it can make systems that are not only precise but also complex, specifically complex enough to make systems as complex as itself.
Now, it further turns out that nanoscale machines can in principle move very fast, and they are by definition small, so it wouldn’t take long for such a nanoscale factory to make all its own components and put them together into another such factory, leading to exponential scaling of productive capacity. Importantly, researchers proposing such concepts are quick to point out that this needn’t be an autonomous, free-range self-replicating system like a cell is — it could be a localized factory model that requires certain specific feedstocks and external controls.
At some level, as synthetic biology matures, for example by incorporating unnatural amino acids into protein chains, or going to cell free systems, there could be significant overlap between a mature synthetic biology and this concept of nanotechnology, particularly in its early development path. But the concept of nanotechnology used here is more general, and it allows the potential use of partially or totally non-biological paths to get there.
So to summarize, over the long term, a nanotechnology worth its salt would:
- Make diverse atomically precise structures, in a programmable way without the need for human intervention, including structures that are well beyond the kinds of sizes and structures accessible to conventional synthetic organic chemistry
- As a note, here is the most complex atomically precise drug synthesized by conventional synthetic chemistry, apparently — impressive, but nowhere near what biology can do
- In saying “diverse” atomically precise structures, I don’t literally mean any possible chemically stable configuration of atoms, I just mean a large design space, where within that design space you have atomically precise control, as opposed to pushing atoms around in “hordes”, i.e., more like constructing with tinker-toys than like making a rice pudding
- Make atomically precise structures that are outside the range of what biology produces, e.g., biology relies on folding of biopolymers to make nanomachines, rather than 2D or 3D printer style “direct write”
- The 2007 Battelle roadmap states this well

- We can summarize this by saying that, in at least some forms, positional chemistry is meant to be “like the ribosome, but for building 2D or 3D covalently linked structures rather than 1D chains”
- Ultimately, go beyond the kinds of environments and physical property ranges accessible to biology, e.g., ultimately could operate in vacuum or at cryogenic temperatures, or temperatures that would turn biological proteins into scrambled eggs — this could be useful if one was aiming to make structures with materials properties (mechanical, electronic, optical) that are well outside those accessible to biology.
- For example, think about the different kinds of brain-like computers one could make that would not be limited by chemical diffusion rates and so on in the ways biological computing is. Or, imagine mechanical devices that sequester CO2 for direct air capture molecule by molecule using motor or ratchet mechanisms, not just by controllable adhesion.
- Ultimately, make nanoscale atomically precise machinery that can itself be directed to carry out such atomically precise fabrication/production processes
- Ultimately, operate at high speeds and in ways that could enable exponential scaling of manufacturing capacity
- Thereby both allow rapid exploration of a much wider range of atomically precise machines and structures than we can today, and also in at least some cases allow massive, fast and cheap production of such machines and structures — think about a shipping container (on Mars if you want) hooked up to some vats of chemicals that becomes a factory for exascale laptops or direct air capture machines operating at the thermodynamic efficiency limit, or something like that [both of these are quite far off indeed at this point, to be absolutely clear]
Again, what I want to talk about here is not the long term, but rather an area of current frontier research, the near-term technical possibilities and activities within it, and what fraction of those we are currently aggressively exploring versus not. I’m not even trying to say that this area of R&D is more important than others at present — just that we should have a reasonably precise roadmap for where it can go and how, and think about how we’re allocating resources to it!
Questioning the positional chemistry concept
Anyway, around the start of the NNI, some were concerned that all this vision was too far out, and insufficiently linked with pragmatic nanoscale research building on, for example, nanoscale materials chemistry as it existed in labs circa the early 2000s. Indeed, it was unclear to many whether this was a sci-fi concept driven by non-technical enthusiasts, or a roadmap grounded in a concrete and defensible technical analysis and experimental plan. In fact, it was best understood as something like both, and neither, but when big funding allocations were on the line, major players in the community faced a binary choice as to how to portray it.
Again, the key idea that the early proponents were pushing for is that of “mechanosynthesis”, which a recent patent (which we’ll discuss more later) defines as:
“‘Mechanosynthesis’ is the use of positional control and mechanical force to facilitate site-specific chemical reactions involved in the building, alteration, or disassembly of a workpiece… Chemical reactions driven by the application of mechanical force include reactions that are (1) driven through its reaction barrier by mechanically forcing reactants or products through the transition state, or (2) driven away from an undesired reaction by mechanically restraining potentially reactive sites from attaining closer physical proximity, or (3) allowed to occur by bringing potentially reactive sites into closer physical proximity when zero mechanical force is required to do so, as for example when no reaction barrier exists, or when thermal energy alone is sufficient to surmount the reaction barrier…”
If one wanted to distance an initiative from enthusiasts getting ahead of themselves, positional chemistry would have been a key concept to discredit. If one disavows the possibility of any kind of generalized, programmable form of positional chemistry, one can still reap the fruits of complex nanoscale devices made by traditional chemical synthesis and self-assembly, but one doesn’t have to deal with the idea of anything approaching a programmable “universal molecular assembler” or something of that sort.
[Michael Nielsen points out that claims about positional chemistry could have been more specific than this, still, and certainly could have been defined and articulated much more clearly than they were at the time. Compare the above narrative to the crisp claim that underpinned the idea of quantum computing, a field which saw sustained growth and legitimacy over the same time period despite also being an initially far-out technical idea: as Deutsch put it in his 1985 paper, “every finitely realizable physical system can be perfectly simulated by a universal model computing machine operating by finite means”. If you want your new field to be based on a stunning level of conceptual clarity and simplicity, look no further than David Deutsch! Alas, Constructor Theory isn’t there yet — or perhaps theoretical physicists are simply a more friendly audience for such ideas than experimental chemists and biologists.]
One particularly aggressive illustration of this conflict was in the so-called “Drexler-Smalley Debate”, which took place in 2001 and 2003 in the popular magazines Scientific American and Chemical and Engineering News (not peer-reviewed academic journals), in which Nobel laureate Richard Smalley lampooned the technical feasibility positional chemistry. It is worth reading the Wikipedia article and the debate itself.
In short, it doesn’t resolve much of anything. Smalley’s initial attack was on a very particular “straw man” notion of positional chemistry, focusing on the impossibility of manipulating each of the individual atoms involved in a bi-molecular reaction individually. But there are many other ways to imagine driving reactions in a positionally-selective way without needing to do this. Perhaps the simplest would be to use the idea of “effective concentration enhancement”, which we will explain below in reference to this 1991 paper, and which is what item (3) in the above definition of positional chemistry also refers to: “reactions… (3) allowed to occur by bringing potentially reactive sites into closer physical proximity… when thermal energy alone is sufficient to surmount the reaction barrier…”. So, Smalley was setting up a straw man.
In a bit more detail, here was Smalley’s main technical claim

and here were the two key replies (per Wikipedia), one from the existence of the ribosome, and one that the argument is a straw man:


Smalley doesn’t address the notion of positional chemistry with any degree of rigor or generality. At the same time, no formal or even particularly concrete specification is given in this discussion for what actually could be achieved with the forms of positional chemistry that would be possible. Unfortunately, the debate kind of derails from there, branching out into a number of other topics but never returning to go deep on these technical issues. So we don’t end up with a technical analysis that would have passed muster among, say, serious physical organic chemists.
As a result of this interchange and related social dynamics, it seems, Drexler was effectively discredited in mainstream academic chemistry circles and materials science circles, and, arguably, it hasn’t been sociologically possible to return even to a serious analysis of this issue since, in the mainstream literature. Smalley himself died just two years later. There are some interesting potential twists on this story here.
Drexler, in a 2004 essay called “Nanotechnology: From Feynman to Funding”, responded by characterizing NNI as implicitly only funding work towards what, in Roco’s above figure, would be called 1st generation “passive nanostructures”, rather than any systematic directed work towards programmable, positionally-directed construction of complex covalently bonded structures and machines. He wrote that “a vastly broadened definition of nanotechnology (including any technology with nanoscale features) enabled specialists from diverse fields to infuse unrelated research with the Feynman mystique… leaders of a funding coalition have attempted to narrow nanotechnology to exclude one area of nanoscale technology–the Feynman vision itself”, and

He then points out that Smalley’s critique can be seen as motivated by a desire to diffuse a “threat to funding” that arose when, for example, Sun Microsystems CEO Bill Joy wrote, in a famous article in 2000 in Wired, that self-replicating nano-robots would pose an existential threat to humanity. [This took quite seriously an idea of autonomous, free-range self-replicating nano-robots that, this essay points out, is arguably an unlikely edge case of a long-term future nanotechnology — it would be much easier and more practical to make nano-manufacturing systems that depend on external control and on specific purified feedstocks, rather than organism-like replicators that would survive in the wild and eat dirt, or what have you. On the other hand, of course, catastrophic risk from biological (rather than nanotechnological) engineering as well as natural biological organisms seems palpable as we speak.]
A possible counterpoint is that funders also wanted to focus on unique/novel material “properties” at the nanoscale, something which is usually heavily related to the quantum mechanics of electrons in materials and under confinement, and which Drexler largely ignores and/or designs around, instead being concerned almost entirely with achieving arbitrary and programmable positioning of the nuclei, caring about the electrons only inasmuch as they influence which bonds can be formed where. Drexler would likely consider this fun science, but not the engineering pie of maximal interest. But many scientists will think otherwise, as would, e.g., the photonics, nanoelectronics, sensors or quantum computing communities. Feynman, on the other hand, did not ignore this aspect in Plenty of Room at the Bottom or in his other writings, e.g., in his lectures on computation he invented a form of electron spin based computing. It is important here to distinguish, also, between the method of fabrication, and the properties of what is being fabricated — what I’m discussing here is almost entirely about the method of fabrication.
Richard Jones has had a lot to say about the situation. Jones argues for a nanotech strategy based more closely on how biology works: “Do the proposals set out in Drexler’s book Nanosystems offer the only way to achieve such a radical nanotechnology?
Obviously not, since cell biology constitutes one radical nanotechnology that is quite different in its design principles to the scaled-down mechanical engineering that underlies Drexler’s vision of “molecular nanotechnology”, or MNT. One can imagine an artificial nanotechnology that uses some of the same operating principles and design philosophy as cell biology, but executes them in synthetic materials (as discussed in Soft Machines). Undoubtedly other approaches to radical nanotechnology that have not yet been conceived could work too. In comparing different potential approaches, we need to assess both how easy in practise it is going to be to implement them, and what their ultimate capabilities are likely to be.”
Certainly progress is being made along those lines in fields that deal with biomolecular self-assembly, synthetic biology and protein engineering. This is indeed exciting and there are likely many flavors of nanotechnology and bio molecular engineering to look forward to— at some level, that’s the “answer” here, i.e., nanotechnology is broad enough and there are enough ways to invent “advanced” forms of nanotechnology that any particular system or capability is ultimately only a small part of the picture. But it seems like Jones’s points are more a question of emphasis, and it doesn’t seem that they actually address the core technical questions in this particular (albeit limited) context about whether one could achieve useful and generalized forms of “positional chemistry” (even if a bunch of work on other, perhaps more bio-inspired systems would be easier or otherwise preferable to working on this in some ways), nor the longer-term questions about what paths the technology could take downstream, although he does have a list of more pointed questions about Drexler’s book Nanosystems here. Jones is pretty upfront about his overall stance here.
What’s Jones’s take on the outcome of the Drexler Smalley debate then? Quoting this OpenPhil report:
“The most high profile opponent of Drexlerian nanotechnology (MNT) is certainly Richard Smalley; he’s a brilliant chemist who commands a great deal of attention because of his Nobel prize, and his polemics are certainly entertainingly written. He has a handy way with a soundbite, too, and his phrases ‘fat fingers’ and ‘sticky fingers’ have become a shorthand expression of the scientific case against MNT. On the other hand, as I discussed below in the context of the Betterhumans article, I don’t think that the now-famous exchange between Smalley and Drexler delivered the killer blow against MNT that sceptics were hoping for.”
Jones 2004, Did Smalley deliver a killer blow to Drexlerian MNT?.
Likewise we have Philip Moriarty’s take: “But I want to take this opportunity to give credit to Drexler. He has been the subject of a lot of criticism – some of it rather non-scientific and ad hominem- from what might be described as the ‘traditional’ (i.e. non-molecular manufacturing) nanoscience community. Drexler deserves significant kudos for the concept at the heart of the molecular manufacturing scheme; single atom chemistry driven purely by (chemo)mechanical forces is demonstrably valid. Richard Smalley, despite raising other important criticisms of the molecular manufacturing concept, misunderstood key aspects of mechanosynthesis and put forward flawed objections to the physical chemistry underlying Drexler’s proposals.”
So the Smalley objections to positional chemistry, per se, clearly aren’t taken all that seriously by physicists who have studied them.
A locus of more useful technical criticism was this series of emails from Richard Jones’s blog, featuring Philip Moriarty attempting to drill into technical details, primarily around a particular proposed scanning probe microscope based scheme, in debate with Chris Phoenix. Jones and Philip Moriarty have helped bring some of the discussion back in a more technical direction and the exchange is very much worth reading. (As a tangent, note the presence of Hal Finney, more known for his role in crypto-currency, in some of the discussions here.) A subset of the discussion (see the exchange with Finney here) had to do with a variant of the Smalley “fat fingers” problem, but most of it had to do with other issues, including
- Specific problems with one particular scheme for scanning probe based mechanosynthesis that is meant to start from current scanning probe microscope technology
- Surface physics, and particularly surface reconstruction re-shaping the nanoscale geometries of probe tips, as a general obstacle, and over what timescales it would have to operate
- Critique of particular schemes for fabricating tips and “handles”
- What has been done in the scanning probe based atom manipulation field thus far and what it does or doesn’t show
- Touching on but not resolving some broader issues, such as how specialized versus universal a mechanosynthesis method can be, the philosophy of different approaches to science and engineering, and definitional quibbling
Because this exchange (admirably) drives into so much detail on particular issues surrounding a particular scanning probe microscope based scheme, though, it doesn’t broadly rule in or out positional chemistry overall, nor even scanning probe based approaches to it.
Was there a concrete experimental proposal for positional chemistry?
It should be clear from the above that there has never really been a serious mainstream exercise to broadly understand possible implementation paths for the “Feynman vision” of using programmable mechanical positioning, like in a 3D printer, to construct of complex objects by controlling the formation or lack thereof of specific covalent bonds, i.e., positional chemistry. Of course, there has been the IBM work on using scanning tunneling microscopes to move single atoms and bond them to specific sites on a surface. But this leaves open most of the key questions about the generality that can be expected of a positional chemistry approach, and of how to achieve a more general positional chemistry capability, e.g., 3D rather than 2D structures, incorporation of larger molecules, or the ability to operate in water as biological ribosomes do. But has there been any concrete analysis of how to actually get there, step by step?
It turns out that circa around 1991, Drexler and Foster (from IBM Almaden research lab, and a lead author on some of the seminal STM based manipulation work) offered at least one concrete proposal, in these three theoretical papers, one of which was published in Nature:
https://www.nature.com/articles/343600b0
https://avs.scitation.org/doi/abs/10.1116/1.585204
https://iopscience.iop.org/article/10.1088/0957-4484/2/3/002/pdf
These were concrete proposals, but not ever followed up on experimentally, as far as I can tell from the published record.
They focused on combining a few known/established abilities into a single system, to demonstrate basic positional chemistry principles in the near term. The known abilities were:
- The ability of scanning probe microscopes (SPMs), such as atomic force microscopes (AFMs), to position a mechanical “tip” to nanometer or sub-nanometer precision, as used in the IBM scanning tunneling microscope atom manipulation work, but here using the fact that AFMs can do this even when the tips are immersed inside water
- The ability of certain macromolecules such as antibodies to bind smaller molecules tightly but reversibly, effectively grabbing them out of solution and holding onto them for a while, and to do so in a defined orientation of capture (this is now famous in 2021, e.g., antibodies binding to specific sites on the SARS-COV-2 spike protein)
- The notion of enhancement of the rate of a chemical reaction by an increase in “effective concentration”: basically, that if one holds one molecule sufficiently near a partner molecule to which it can react, rather than letting it roam free throughout the entire test tube, the reaction will happen much faster, since thermal jiggling motions have a much higher chance of causing the two reactants to collide in a favorable orientation to allow the reaction to proceed (i.e., to cross the necessary “transition state”)
- Further, the fact that this type of enhancement can be highly spatially selective, such that if the molecule were held in place just a nanometer or two further away from its partner, the effective concentration enhancement of the reaction would drop precipitously — this is basically what is calculated in the Nature paper
- Note that this idea is the same as item (3) in the three types of mechano-synthetic guidance quoted above. Again, “reactions… (3) allowed to occur by bringing potentially reactive sites into closer physical proximity… when thermal energy alone is sufficient to surmount the reaction barrier…”.
So what Drexler and Foster propose to do is to attach a macromolecular binder like an antibody to an AFM tip, and to place it in a solution containing many copies of a molecule which we would like to add to a mechano-synthetic workpiece. The antibody on the AFM tip would then grab one of these molecules from solution (call it Molecule1GrabbedFromSolution), and hang onto it for a while. Then, using the AFM’s precise mechanical positioning ability, it would bring Molecule1GrabbedFromSolution within a nanometer or so of a potential partner molecule (call it Partner1) to which it could react, located at a specific position on the workpiece. If the nearest adjacent binding site to a potential partner molecule on the workpiece (call it Partner2) is located a couple of nanometers away, then we can expect it to react much, much faster with Partner1 than Partner2, because of the effective concentration enhancement, especially if the background concentration of molecules floating in the solution is low enough.
Thus, with high probability, we’ll have site-selectively reacted Molecule1GrabbedFromSolution with Partner1 versus with Partner2 who is located only a short distance away. Once that site-selective covalent bond is formed, we can pull back the AFM tip, wash out the current solution of molecules and wash in a new and potentially distinct type, and then grab one and put it in another site-selective reaction at a different, user-defined site on the growing workpiece.
This would be a slow and cumbersome but fundamentally valid form of positional chemistry, which could form a wide variety of reactions depending on how one chose the identities of the molecules on the workpiece and those to wash into solution, and which would operate in water at room temperature and therefore be compatible with any number of known biomolecular reaction chemistries in particular cases. This is unlike IBM’s moving atoms with the STM, which are in high vacuum, and hence very restricted in which kinds of building blocks they can use. It also means that one could use reactions one knows to work in solution, albeit at slow rates, using effective concentration enhancement to boost those rates — this means that complex quantum mechanical modeling of chemical intermediates or side reactions and so on would not be required, as one could use a robust toolkit of already-known, off-the-shelf reactions.
Drexler apparently intended this as a stopgap demo, to be done before using a self-assembled covalent molecular “printer” instead of a scanning probe microscope for positional control, per his 1979 idea of protein-based machines making other molecular machines.
In Chapter 5 of the 1991 book Unbounding the Future, the authors suggest as a plausible development mode for the above project: something like a DARPA program, namely, a directed collaboration of labs with expertise in supramolecular chemistry, atomic force microscopy, surface chemistry, antibodies, and so on. This would be the interface of the “proximal probe systems” (AFM) and “chemistry, protein engineering” (antibodies, appropriate choices of reactant partners, surface passivations, and workpiece anchorings) communities. They then suggest that this could bootstrap towards more advanced systems:
Here are some pictures of this idea. First, the antibody-like gripper on an AFM tip, grabbing a smaller, reactive (but not too reactive) molecule from a liquid solution, and hanging onto it for a while, before moving into position to add it to a workpiece at a user-defined location

That’s the conceptual schematic from the popular book. In the academic papers, the situation is slightly inverted in that the tool holder (antibody) and reactive molecular tool are to be localized on a flat surface, with the AFM tip actually moving around a bead containing the growing molecular workpiece. Anyway, here it is zoomed in, and then slightly out

and then zoomed out further showing beads on the AFM tip, only one of which comes close to the flat surface that presents the molecular tool
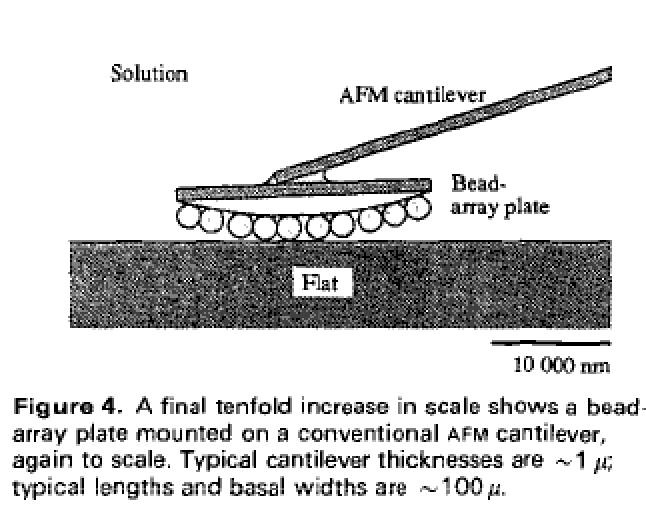
Finally they proposed some chemistry for attaching the antibodies on the flat surface in a reasonable orientation and with enough spacing from one another that only one would be within reachable distance of a workpiece on the closest part of the closest bead
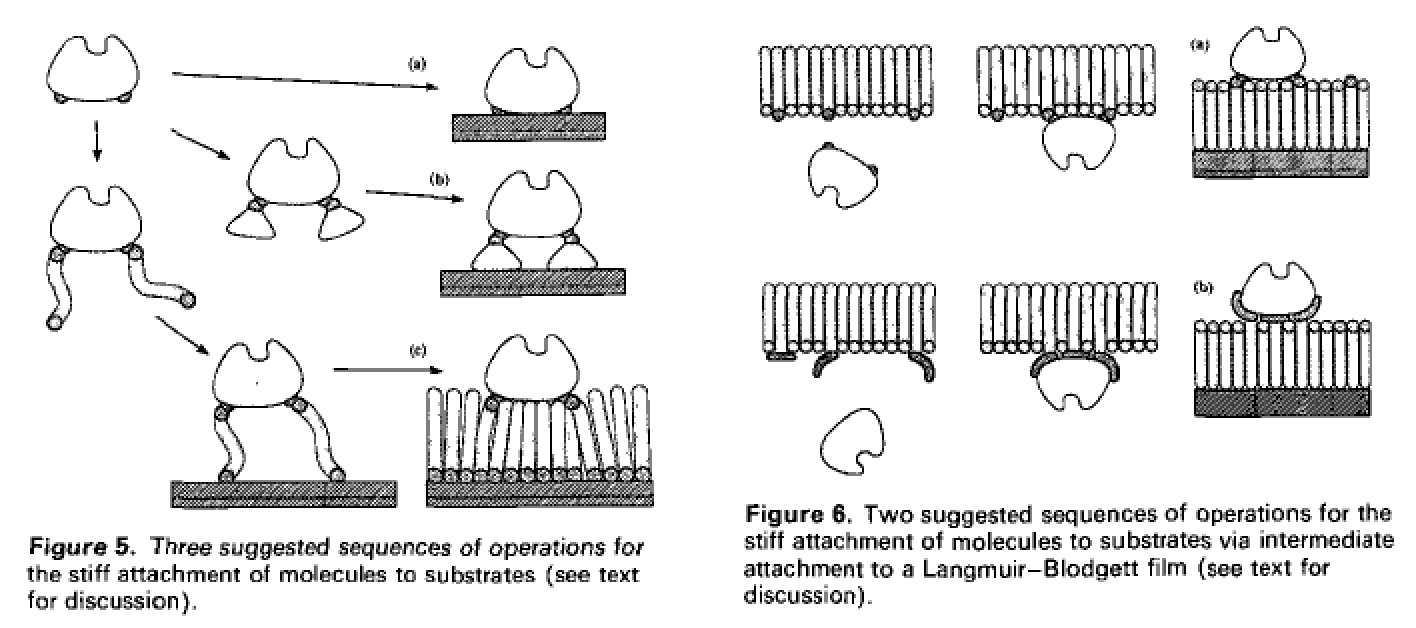
So that’s a heck of a lot more concrete and physically well justified than any “straw man” (e.g., of directly manipulating all individual atoms in the two molecules-to-be-reacted at the same time) that Smalley critiqued a decade later.
Here is a quote from one of the papers, which hopefully is understandable now given the context just provided

[Has anyone done anything like this, in the 30 years since? Well, there is certainly work on measuring antibody antigen binding at the single molecule level with the atomic force microscope, and indeed a whole field of AFM based single molecule force spectroscopy for scientific studies of biological proteins. Ebner’s lab has put both antibodies and DNA tetrahedra on AFM tips. Then there is something called “single molecule cut and paste surface assembly” which uses a DNA molecule attached to an AFM tip to pick up and move other DNA molecules, eventually dropping them down at a desired location where they can bind to DNA on a surface. This is getting close, but doesn’t drive covalent chemistry, and the relevant DNA strands are relatively long and floppy compared to the 1991-proposed scheme. So, in short, no.]
[It is also worth noting that the idea of effective concentration enhancement and the use of a form of programmable positioning to achieve it has also been utilized in a much more mainstream field, that of DNA templated organic synthesis. In this case, the programmable positioning is by using a DNA strand as a kind of “necklace” for bringing together various chemical “jewels” (molecules A-F just below) to react in certain combinations:
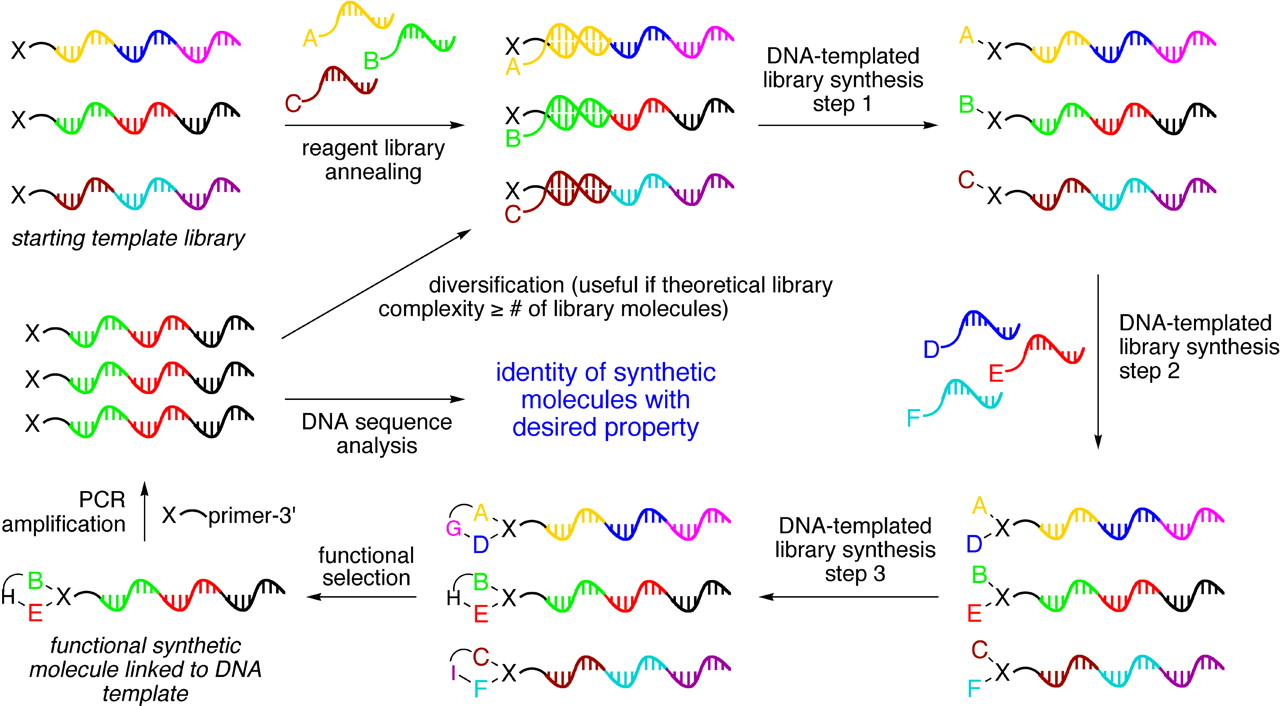
One take-home message here is that these are worthwhile goals, but you have to be ruthlessly concrete and focused on experimental demonstrations to get real buy-in for such goals from experimental chemists and biochemists — the abstract vision alone, even if appealing to engineers and computer scientists, doesn’t get sufficiently robust traction in the empirically driven realms of chemistry and biology. Even this relatively-concrete paper wasn’t concrete enough, apparently.
Another take home message, though, is that it is plausible that there has been a sociological bottleneck, whereby it has basically become taboo to try to work too explicitly on positional chemistry, following the Drexler-Smalley debate and associated events. Because positional chemistry is also a very hard systems problem, beyond the reach of any one scientist or lab, individual courageous researchers can’t escape this sociological trap by quietly showing a demo. We’ll come back to this later, but arguably the theoretical cases for various forms of positional chemistry being possible with modest advances on existing technology are pretty strong, and as we’ve seen above and will see more below, there have been various decently concrete experimental sketches, but these have not seen the kind of funding or concerted, explicit team efforts that we see in other forward-looking fields like quantum computing.
Building blocks that emerged in the meantime
We’ve just learned that there were at least the beginnings of at least one directed/roadmapped proposal for getting started in demonstrating positional chemistry ideas even circa the early 1990s. More generally, we’ll see that ideas close to the “Feynman vision” — of positionally directed assembly involving site-specific formation of certain covalent bonds at the expense of otherwise chemically equivalent bonds with their neighbors — have bubbled up several times. But this hasn’t been the main focus of any major funding pushes (until around 2018-2019, per both a small DOE program, and a secretive Canadian company, as we’ll see below, although Philip Moriarty had received at least one small grant circa 2007 to do experimental work on the molecular manufacturing theme), or of most anyone’s serious and sustained experimental work over that intervening period. [Per the article on the 2007 grant, the Moriarty grant was with Rasmita Raval. Andrew Turberfield also had a DNA-based “artificial ribosome” direction as part of the project, and has a new grant on that now
https://gow.epsrc.ukri.org/NGBOViewGrant.aspx?GrantRef=EP/T000562/1
All of this is in the UK.
“The ambitious projects, which are funded to 2010, were hammered out in January at a hotel outside Southampton, where scientists and EPSRC members took part in an “Ideas Factory” workshop, giving them free rein to discuss bold ideas which might not have been funded under the usual peer-reviewed grant calls.” Total appears to be <$1M/year across the projects.]
Regardless, a lot has happened in nanotechnology since 1991 that, though not explicitly directed along this “positional covalent assembly” path, is nonetheless extremely relevant to how one should think about such problems today, and/or is simply compelling in the broader realm of technology platforms for “programmable atomically precise fabrication”. I’ll go through some of this quickly to show where we’re at now.
This is not a core logical part of the feasibility argument around positional chemistry that we’re making — that’s more about the theory and about sociology. It is more an illustration of primitives that have arisen in the last 20 years or so that make the playing field more interesting overall.
I’ll focus only on a few key advances here that are most relevant to possible ways of bringing forth positional chemistry, and relegate others to an Appendix.
DNA nanotechnology
The 2010 Kavli Prize in nanotechnology was awarded to Don Eigler, of the IBM STM atom manipulation work, and Ned Seeman, “for their development of unprecedented methods to control matter on the nanoscale”. Seeman was the key early developer of DNA nanotechnology.
The basic idea of DNA nanotechnology is that the specificity of Watson-Crick DNA base pairs (A pairs with T and C pairs with G across the two strands of the famous double helix) make it very easy to rationally “program” the self-assembly of single-stranded DNAs (one strand of the helix) into user-defined geometric structures. Here is Seeman’s delightful example of a cube
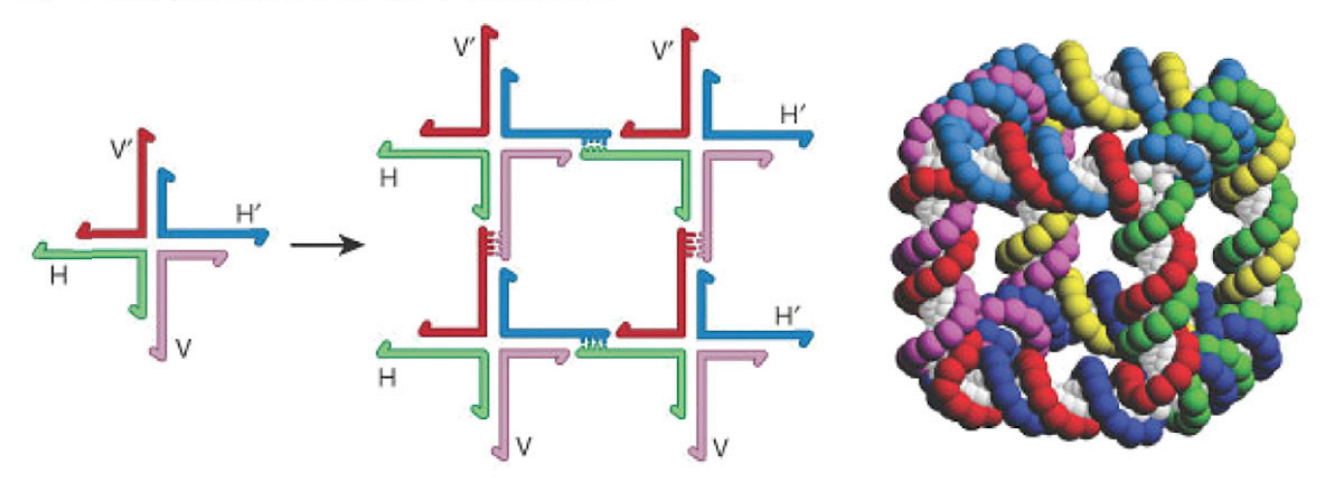
On the left the individual lines are single short chains of DNA (half of the helix). Their sequence complementarity is such that they create a four armed junction which in turn can assemble into the cube just by random collisions in solution.
The next iteration on this used a different kind of junction called a double crossover, inspired by a structure formed during DNA recombination inside cells
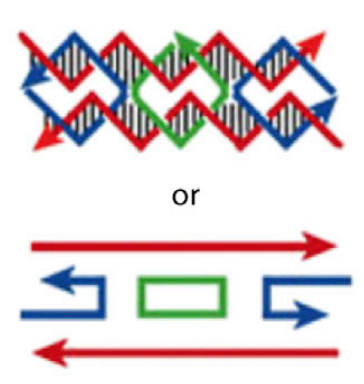
The thin black vertical lines in the top half of this last figure all represent individual Watson-Crick base pairs, A::T and C::G.
The great thing about this is there is no protein folding problem involved, and no bespoke chemistry involved — it is a very simple, facile design process, where you need only to know (more or less) about the periodicity of the helix, and complementarity of A with T and C with G.
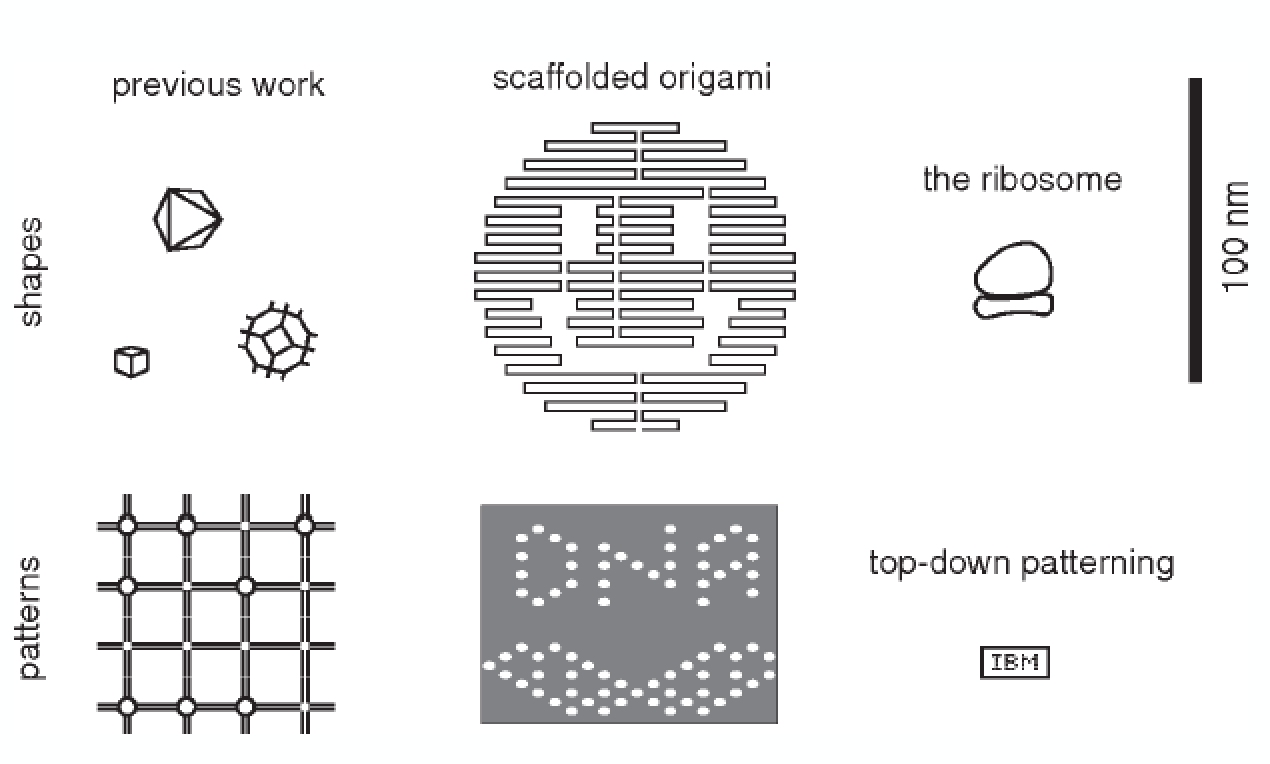
DNA origami
To my mind, the big breakthrough in DNA nanotechnology came in 2006 with the development of “scaffolded DNA origami”. This uses many double crossover structures to link together a bunch of DNA helices in parallel arrays, to form complex shapes, which are much larger in size than the above cube, about 100 nm on a side. This works by using lots of short single-stranded DNA “staple” strands to link different parts of a long scaffold strand (from a single-stranded DNA virus that infects bacteria). From Paul Rothemund’s 2006 paper, here the top row are the target designs and the bottom rows show AFM images of the resulting structures after assembly:

You simply add the staples strands, some salt, and the scaffold strand together in solution, heat and slowly cool, and trillions of these self-assemble inside the test tube. Minds were blown, and this was a single-authored paper on the cover of Nature. (I recommend skimming the 82-page supplemental materials for this paper to get a sense of what Paul Rothemund actually did.)
Here is a nice animation of how this works, from Shawn Douglas
Importantly, each short staple strand (and each region of the scaffold strand) has a unique sequence unlike that of any other staple strand, meaning that the structures are “fully addressable” — a given DNA staple strand lands in one and only one place in the structure. By attaching some other chemical to a given DNA staple, you can direct it to its particular location. Thus DNA origami works as a kind of “molecular breadboard” where you can attach what you want where you want, with a precision of a couple nanometers, about the size of one staple strand.
This illustrates the size scales of DNA origami relative to other nanoscale entities, including the ribosome and the IBM atomic patterns
To understand a bit about the sizes, note that the width of a DNA helix is about 2 nm and the spacing from letter to letter along the chain is about 0.34 nm. We can’t get quite that precision of patterning in DNA origami though; quoting from a recent paper one gets more like 5 nm spatial design accuracy (which is to be distinguished from thermal fluctuations around that design)

Anyway, you need hundreds of letters worth of DNA along the horizontal axis and dozens of DNA helices along the vertical axis to get a 100 nm x 100 nm smiley face like the one above. Overall we’re talking on the order of 10,000 DNA base pairs and on the order of a million atoms in such structures (putting a few together we can get giga-dalton structures where a dalton is the weight of one hydrogen atom).
In the 2007-2009 era, William Shih’s lab extended this to 3D DNA origami structures, i.e., rigid 3D assemblies of linked parallel DNA double helices, which are also “fully addressable”, in the sense that a given staple strand is destined for a unique position in the structure, and thus can be attached to some other molecule in order to bring it there
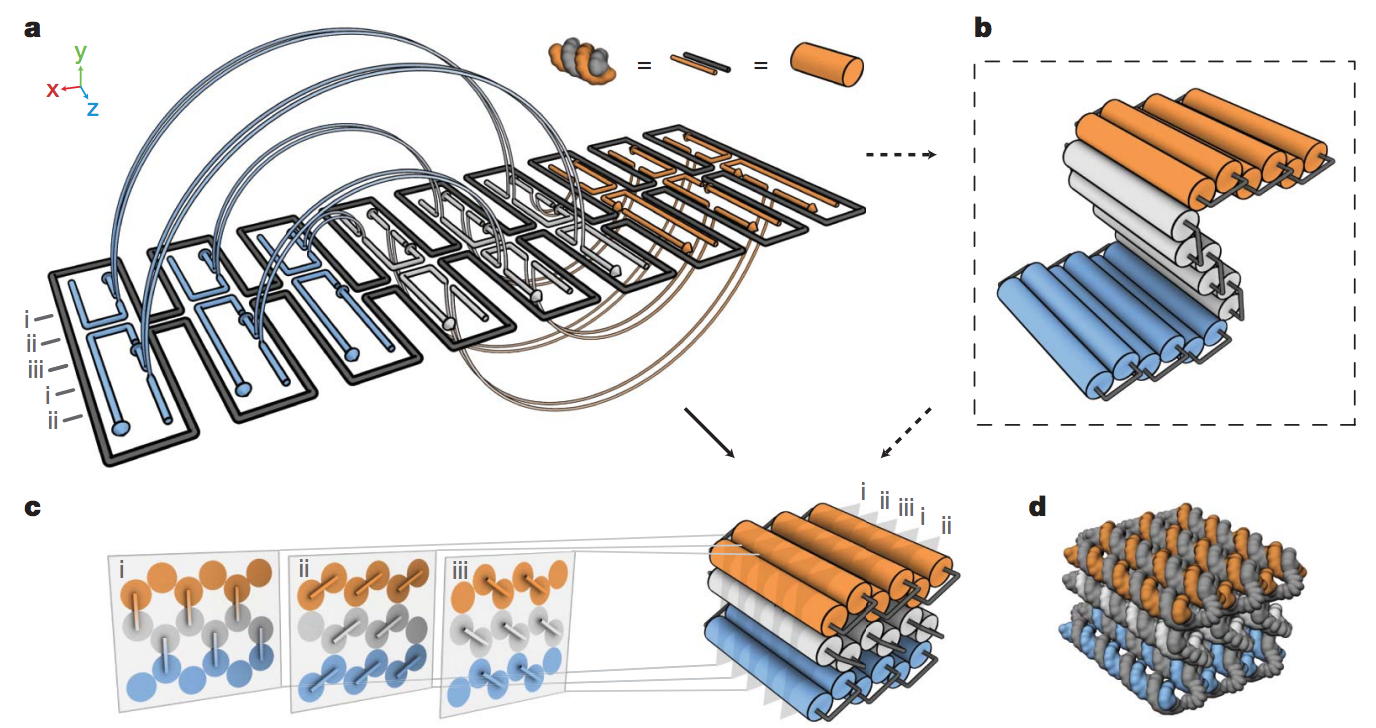
I was the research assistant of his graduate student Shawn Douglas at the time, and we wrote a simple graphical design software to help choose the staple strand sequences for a given target shape, called caDNAno. This in turn leads to 3D mechanical DNA machines made out of the 3D DNA origami: here on the right, each little cylinder represents a DNA double helix
This actually kinda works, as seen in the electron microscope images below

Alas, actuation schemes for such are very crude currently, but Shih lab is working at least in a very preliminary way on a proper stepper motor design using 3D DNA origami, see below; in principle, DNA origami based stepper motors can be actuated by light exposure, pH changes, small molecules and so on. DNA binding and strand displacement reactions on DNA origami have been used to create systems reminiscent of molecular assembly lines, arguably demonstrating a very rudimentary form of non-covalent but still positional nanoscale assembly.
Protein carpentry
Proteins are more complex to engineer than DNA, having complex folds (see here for an explanation) and many kinds of interactions to deal with (hydrophobic, charge, disulfide bonds, secondary structure motifs, more complex patterns of hydrogen bonds, different sizes and shapes of amino acid) beyond just simple pairing rules like DNA has. However, an emerging field of “protein carpentry”, if you will, looks at restricted subsets of proteins with more predictable backbone shapes, and seeks more modular binding interfaces between them.
One such class is coiled coil proteins, and David Baker’s lab has recently designed a set of orthogonal pairs, i.e., a set of coiled coils within which each has only one possible binding partner, much as any short single stranded DNA has a unique complementary strand to which it binds
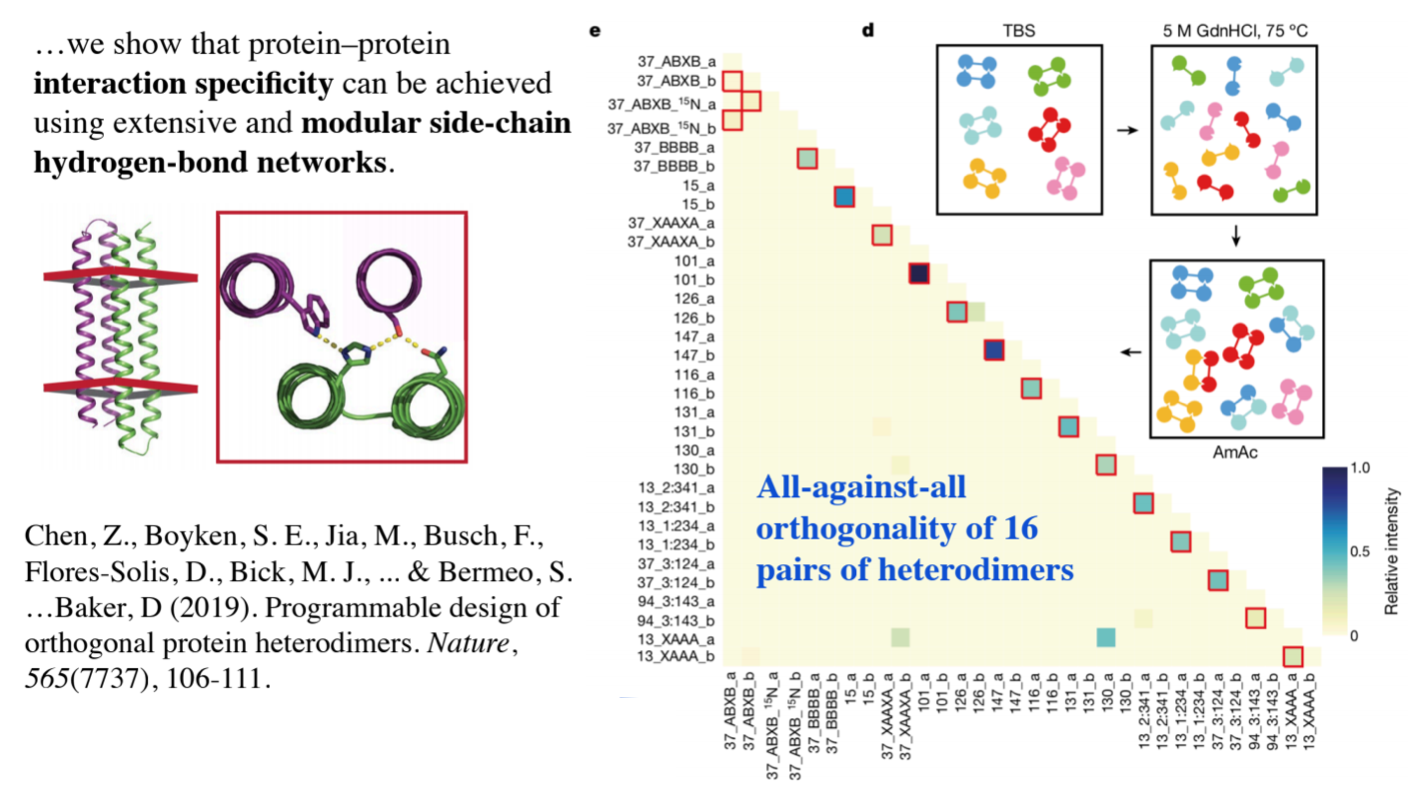
Another family of proteins that could be used for such a purpose would be beta solenoid proteins, which can have flat surfaces like this

which could be decorated with charge or hydrogen bonding patterns that would mediate specific pairwise interactions in the spirit of DNA.
Recently, other work from Daid Baker’s lab is also taking a more modular, mechanical approach to protein based self-assembly, using rigid junctions

(We haven’t mentioned peptoids, as opposed to peptides — a protein is a poly-peptide — here but they are interesting.)
Spiroligomer “molecular lego”
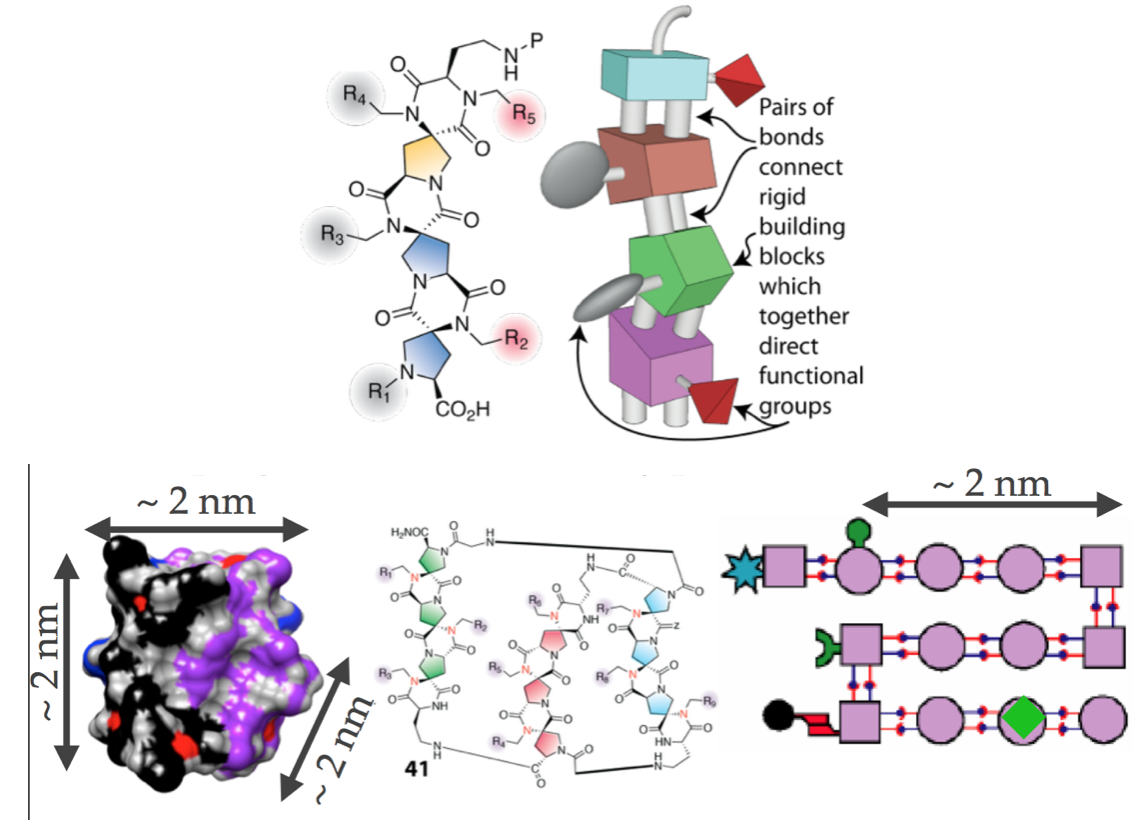
At each unit along a protein chain, there are two flexible angles around which the chain can rotate. This is what leads to protein folding — the ultimate fold is the pattern of such angles it chooses. Christian Schafmeister developed Spiroligomers for more direct rational design of protein structures. Unlike proteins, they lack rotational freedom at each joint, instead locking in specific angles — this works by using pairs of bonds, which one can’t rotate around, to connect adjacent elements along the chain, not just single bonds, which one can rotate around. Spiroligomers are like proteins, but instead of using non-covalent interactions to determine folding (hydrogen bonds, charge, hydrophobic), you directly lock in the final fold based on the covalent structure of the molecule.
Quoting from their papers:
“Proteins attain… well-defined structures through the complex process of protein folding. We… emulate these protein functions by constructing macromolecules that are easier to engineer by avoiding folding altogether… Bis-peptides use the conformational preferences of fused rings, stereochemistry, and strong covalent bonds to define their shape, unlike natural proteins and synthetic foldamers, which depend on noncovalent interactions and an unpredictable folding process to attain structure.”
These approaches can be used to make structures a couple of nanometers on a side with well defined shapes and specific chemical functional groups in well defined positions, and arranged and oriented along multiple axes:
Action-PAINT: single-molecule patterning by a ratchet mechanism
This one takes a bit of work to explain. It is in the general category of nanopatterning via “running a microscope in reverse”. Basically, there is a microscopy method called DNA PAINT that works like this. You have some DNA strands on a surface, arranged just nanometers apart from one another, and you want to see how they are all arranged. If you just put fluorescent labels on all of them at once, and look in an optical microscope, then the limited resolution of the optical microscope — set by the wavelength of light, a few hundred nanometers — blurs out your image. But if you can have complementary DNA strands bind and unbind transiently with the strands on the surface, fluorescing only when they bind, and such that at any given time only one is bound, then you can localize each binding event, one at a time, with higher precision than the wavelength (by finding the centroid of a single Gaussian spot at a time). That’s the basic principle of single-molecule localization microscopy, which won a Nobel Prize in 2018
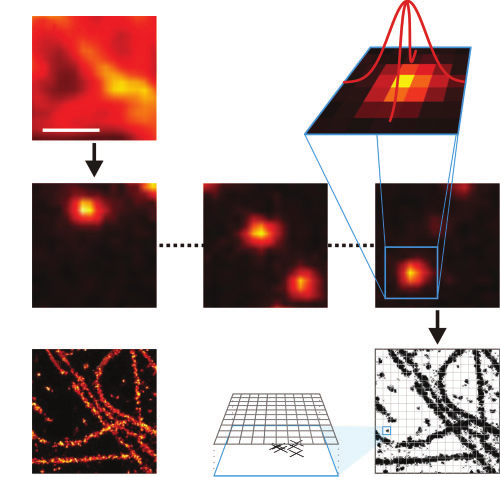
The magic is that you can localize the centroid of one (and only one) isolated fluorescent spot much more precisely than you can discriminate the distance between two (or more) overlapping fluorescent spots. So you rely on having a sparse image at any one time, as DNA molecules bind on and off to different sites on the object such that typically only one site has a bound partner at any given time on, and then you localize each binding event one by one and build up the overall image as a composite of those localizations.
Anyway, that’s a microscopy method that lets you see with resolution down to a couple nanometers, well below the wavelength of light.
How can you use this for nano-patterning? Well, imagine you have a desired pattern you want to make, and you are doing this “single molecule localization microscopy” process in real time. Then, if you can detect that a DNA strand has bound to a spot that is supposed to be part of your pattern, and you can register this in real time, then you can quickly blast the sample with a burst of UV light which locks that strand in place, preventing it from ever leaving again. That “locks in” a DNA bound to that spot. Now, most of the time, the localizations you’ll see will be at spots you don’t want to be part of your pattern, so you don’t blast the UV light then. But every so often, you’ll see a probe bound at a spot you want to be in the pattern, and when that happens, you take fast action, locking it in. That’s what ACTION-PAINT does:
This can be seen as a kind of molecular printer with in principle roughly the same resolution as that of the underlying single molecular localization microscopy method. Which in practice is not quite as high as the best AFM positioning resolution. But it is pretty high, in the single digit nanometers in the very best case.
Scanning probe based surface atom manipulation
We covered this a bit above in relation to Eigler and IBM. By 2002, Eigler’s group had even made patterns of atoms that could create little logic gates that functioned based on tiny molecules hopping between adjacent binding sites on a surface
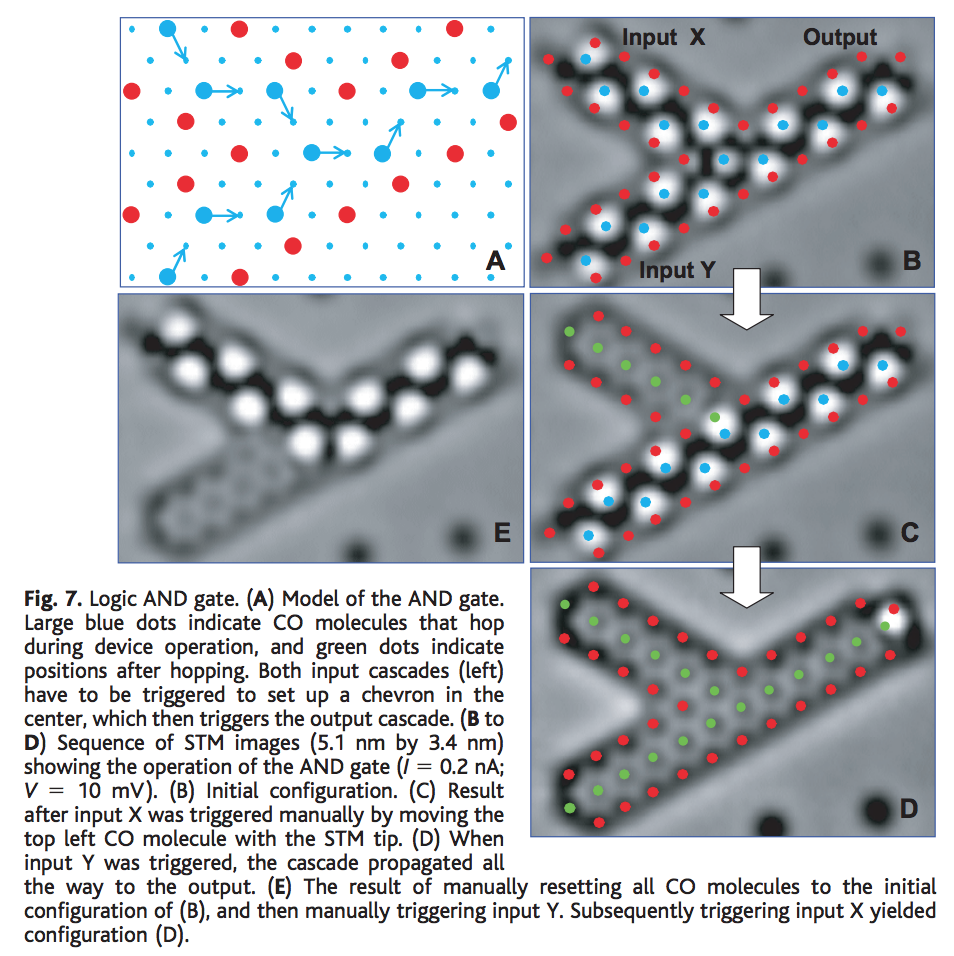
IBM continued this line of work and by 2013 had some cool atomic stop-motion movies to show
There has also been single atom level mechanical manipulation and patterning using AFMs rather than STMs, and using very fine electron beams.
One cool recent paper used voltage to control an individual chemical bond’s strength
https://physics.aps.org/articles/v14/s64
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.126.216801
This could be used as a way to control the “stickiness” of scanning probe based atomic manipulation tools, potentially. [Thanks to Jacob Swett for this comment.]
Finally this has been used in a non-scaled, proof of concept way in the context of silicon lithographic chip making. Quoting from a NAS report:
“An explicitly lithographic process with atomic site specificity is the “hydrogen passivation resist” pioneered by Lyding’s group at the University of Illinois in the 1990s. The process involves covering (passivating) a silicon wafer with a single layer of hydrogen atoms and removing selected hydrogen atoms with an electrical current from a scanning probe tip. The hydrogen-silicon bond is stable enough that the resulting pattern can be used to mask further chemical reactions on the surface, with atomic site specificity, at room temperature and above. In 2004, scientists associated with the Australian National Quantum Computer project used this method to introduce single atoms of phosphorus into a silicon crystal at selected atomic sites.”
My take home message, from this section overall, is that there has actually been lots of progress in programmable, directed nanoscale patterning methods, using new thinking beyond standard organic chemistry or bulk materials science methods — nanotechnology has been exploring not just nanoparticles, thin films & general materials science, but also interesting new ways to assemble complex systems in directed ways.
But how does this stack up against the “Feynman vision”? We have interesting nanoscale patterns and some interesting nanoscale machines and devices, but not machines directing complex covalent chemistry (which bonds occur where) positionally
Gestures at explicit progress on positional chemistry
Two paths within positional chemistry
To be clear, there are really two possible work paths within “positional chemistry” at this point.
One goes directly for building dense covalent diamondoid structures using specialized mechanically controlled reactions in a vacuum chamber, directed using something like a scanning tunneling microscope tip. We’ll call this “mechanosynthesis, in vacuum”.
The other basically takes off the shelf chemical reactions that are known to take place in water, and makes them site-selective on a 2D or 3D workpiece with an array of possible binding/bonding sites separated from their neighbors by a distance larger than the positional accuracy of the best printer we can currently self-assemble in water. This is the “molecular 3d printing” path. We’ll call this “ribosome-like, in solution”.
Conceptually, molecular 3D printing is supposed to be “like the ribosome, but assembling 2D or 3D chemically linked structures, rather than 1D chains (that then fold into 3D)”. The bricks that could be printed would be from some finite set (just as the amino acids linked together by ribosomes form a set of 20), chosen to be big enough and to be compatible with producing the sufficiently-widely-separated binding sites for the next layer of printing.
The sites the bricks bind to (which are displayed from the previous layer of bricks) could be “activatable”, such that the print head of a 3D printer just activates certain sites for binding/bonding, while keeping others chemically protected, but the actual delivery of the bricks they bind to is by free diffusion in solution. Drexler has pointed out that this is a generalization of the protecting group chemistry used to synthesize DNA or peptide chains chemically with control over sequence, to a setting where the deprotection or activations steps are positionally controlled over a 2D or 3D lattice of possible addition sites.
Alternatively the print head could directly transport each brick to its desired binding site. After each round of printing there could also be a “snap together” step where other bonds are formed laterally among the bricks, for example, ratcheting them into the right orientations.
Here is a summary of the differences, and then the histories of these two paths:


Explicit work on positionally directed covalent chemistry
OpenPhil and NAS published analyses
Here is a quote from an Open Philanthropy Project report on (the possibility of risk from) atomically precise manufacturing, summarizing a 2006 US National Academy of Sciences report that briefly evaluated the idea of nanotechnology based positionally directed covalent chemistry, which they referred to as “Technical Feasibility of Site-Specific Chemistry for Large-Scale Manufacturing”

Overall, the NAS report is pretty good and highlights a number of relevant areas for focused research, including Schafmeister’s spiroligomers mentioned above: “Preliminary experimental validation that such nanobiotechnology may be useful for manufacturing is found in the ability to design synthetic bis-amino acid oligomers to have specific rigid shapes, which should be useful in constructing complex atomically precise three-dimensional objects”.
It also has a pretty good description of the positional chemistry idea:
“The proposed manufacturing systems can be viewed as highly miniaturized, highly articulated versions of today’s scanning probe systems, or perhaps as engineered ribosome-like systems designed to assemble a wide range of molecular building blocks in two or three dimensions rather than the linear assembly of amino acids by the ribosome. In this approach, reactions are described with both reagent and product as part of extended “handle” structures, which can be moved mechanically.”
There was also the 2007 roadmap run by Battelle, which administers the National Labs, focused on the notion of atomic precision, but its recommendations remained a bit high-level, articulating design principles but not full designs or experimental plans. It is very much worth reading though, and quite well written. More on it a bit later.
Continuing from the 2006 NAS report:
“Proponents of these design and manufacturing strategies foresee the exploitation of exquisitely controlled site-specific chemistry on a vast industrial scale. While scanning probe systems have demonstrated the feasibility of some site-specific reactions, scale-up to manufacturing systems is still a daunting task, and the majority of nanoscale scientists and engineers believe it is too early to try to predict the ultimate capabilities of such systems… The committee found the evaluation of the feasibility of these ideas to be difficult because of the lack of experimental demonstrations of many of the key underlying concepts.
The technical arguments make use of accepted scientific knowledge but constitute a ‘theoretical analysis demonstrating the possibility of a class of as-yet unrealizable devices.’
…Construction of extended structures with three-dimensional covalent bonding may be easy to conceive and might be readily accomplished, but only by using tools that do not yet exist. In other words, the tool structures and other components cannot yet be built, but they can be computationally modeled. Modeling the thermodynamic stability of a structure (showing that it can, in principle, exist) does not tell one how to build it, and these arguments do not yet constitute a research strategy or a research plan.
To bring this field forward, meaningful connections are needed between the relevant scientific communities. Examples include:
- Delineating desirable research directions not already being pursued by the biochemistry community;
- Defining and focusing on some basic experimental steps that are critical to advancing long-term goals; and
- Outlining some “proof-of-principle” studies that, if successful, would provide knowledge or engineering demonstrations of key principles or components with immediate value.
Research funding that is based on the ability of investigators to produce experimental demonstrations that link to abstract models and guide long-term vision is most appropriate to achieve this goal.”
It is interesting to me that they don’t cite the above-discussed Drexler/Foster Nature correspondence or the follow-on papers proposing the antibody-functionalized surfaces, effective concentration enhancement and AFM manipulation, as a concrete desirable research direction or in regards to “defining” some basic experimental steps.
But anyway, what work has actually been done since that is explicitly on positionally directed chemistry?
Canadian mechanosynthesis effort
Well, it looks like the Candian government is funding some such work, potentially up to the tune of about $200M:


Little is clear about exactly what they are doing or how much progress they have made, but some of the original “Feynman vision” enthusiasts like Ralph Merkle are leading the charge up there at the Canadian Bank Note (CBN) corporation, judging by the authorship on some of their publicly visible patents, like these, with titles like “Systems and methods for mechanosynthesis”
https://patents.justia.com/patent/10822229 (this one especially makes for amusing reading)
https://patents.google.com/patent/US10309985B2/
https://patents.google.com/patent/US8276211B1/
(Merkle is also of the inventors of public key cryptography, whose name has become more familiar again of late because of the role of Merkle trees in cryptocurrency algorithms; incidentally, I find it pretty interesting that a lot of the early developers of these ideas were at major corporate research labs like Xerox PARC or IBM Almaden at the time, or at NASA.)
In short, they are, based on what’s in these patents, pursuing positional diamond mechanosynthesis, initially using various specially designed chemical tools attached to scanning probe microscope tips. Here are two examples of the kind of molecular tip they would use — each meant to facilitate a specific reaction on or near its end-effector
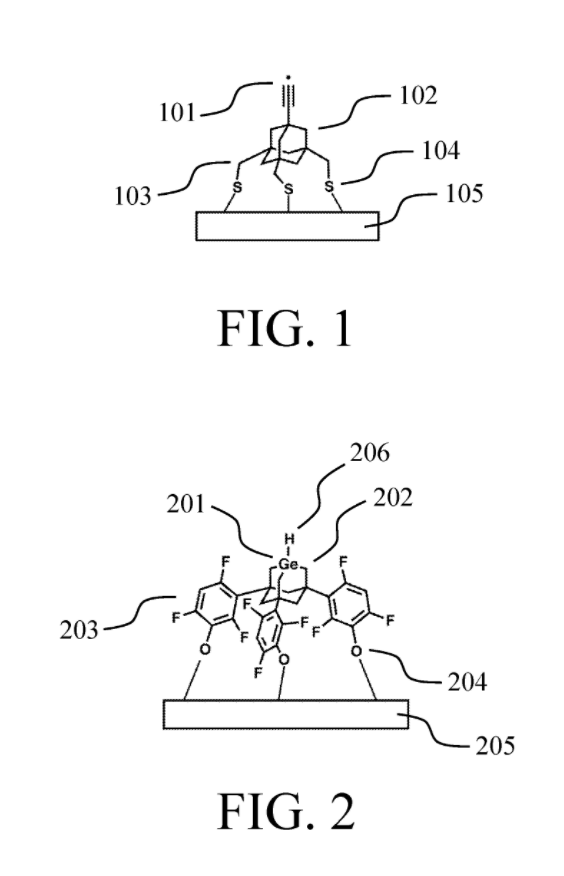
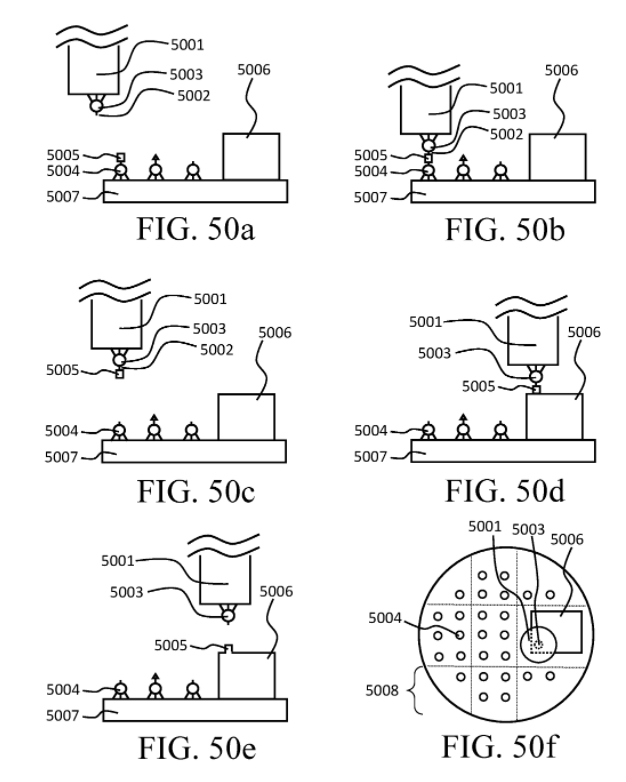
and here is schematically how they would add/remove molecular atoms from a 3D diamondoid workpiece
A lot of this is basically very similar to the stuff they published years before, for instance this 1998 computational study on mechanosynthetic reactions, showing a bit more the chemical transformations they are aiming to drive at specific sites on a workpiece using different kinds of molecular tip
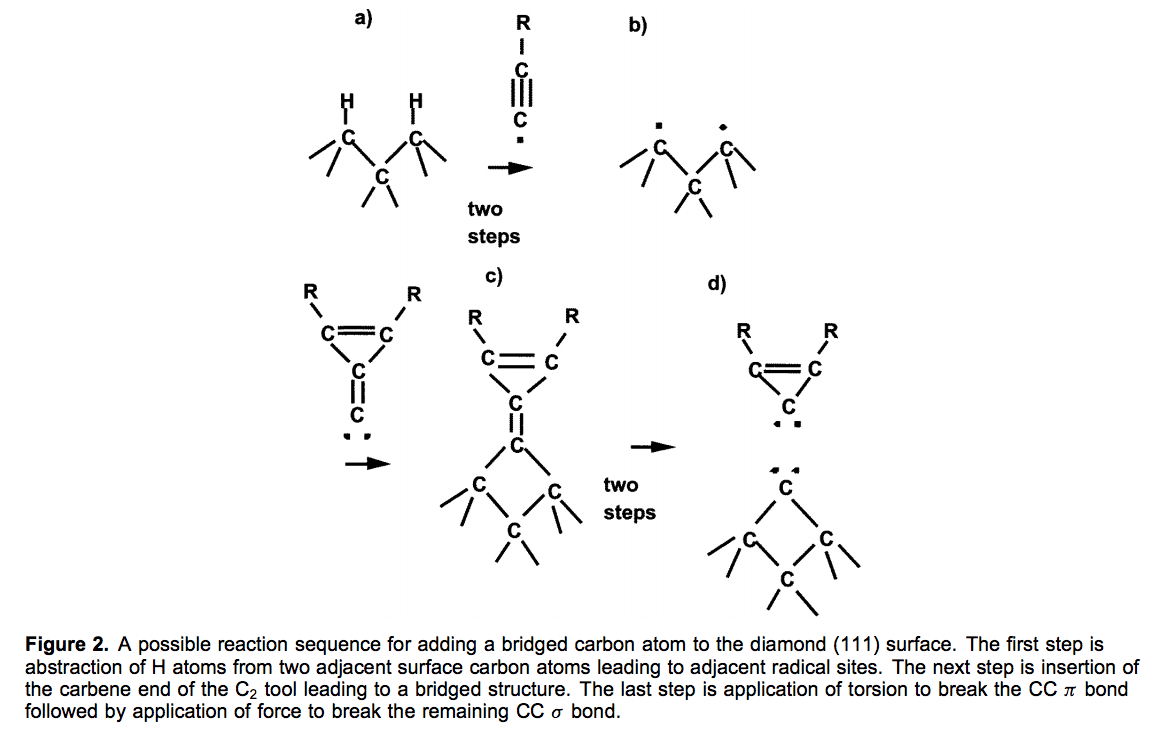
There are approximate first-principles quantum chemistry (usually density functional theory, DFT, using standard Gaussian software) simulations suggesting that such mechanically directed reactions could work:
https://www.ingentaconnect.com/contentone/asp/jctn/2005/00000002/00000001/art00003
https://www.ingentaconnect.com/contentone/asp/jctn/2008/00000005/00000005/art00002
Probably this Canadian entity CBN Nanotechnologies has done many more such simulations by now… but do they have all the experimental kinks worked out? I highly doubt it. They have not published any experimental demos to date. Then one must ask, how could this be made to scale? Their plan would presumably be to use this approach to make smaller diamondoid machines that are themselves STM-like probes/manipulators and then in turn use these to make more. Although IBM has also looked at massively parallel scanning probe systems, one of which is called “the Millipede”.
The CBN work seems to grow organically from Merkle’s earlier work, see
http://www.molecularassembler.com/Nanofactory/AnnBibDMS.htm
Possible challenges
I wish them well, but, for better or worse, as revealed by the above quotes from the NAS report, approximately nobody in mainstream chemistry or biology will be convinced by such simulations and theoretical design work. DFT simulation is a real thing now, it can now begin to accurately simulate liquid water (see the discussion James McBride’s freshman organic chemistry course mentioned in Appendix 1, around self-consistent field calculations of molecular orbitals, to get a quick sense of how such simulations work), but nevertheless — academic chemistry and biology has an empiricist “I’ll believe it when I see it” attitude to many things, perhaps for good (historical and other) reasons.
A key issue for this approach is that the tips are highly reactive. So these reactions must be done in vacuum (or inert gas atmosphere) to prevent side reactions from occuring, and there are a lot of requirements on the cleanness and flatness of the surfaces involved, presumably, and probably the reaction temperatures need to be kept low as well, not to mention all the practicalities of functionalizing probes and surfaces with specific molecules in the right orientations. If you look at the above reaction scheme, too, look at all the fancy mechanical manipulation of the tip they need to do, like “application of torsion to break the C-C pi bond”. This is at best really non-trivial by way of scanning probe manipulation.
So, in some sense, this looks like a serious effort, but also one with numerous potential big and hard to avoid pitfalls. Here was Drexler’s commentary from a discussion with OpenPhil about the disadvantages of this kind of high vacuum scanning probe direct-to-diamondoid approach, versus one that exploits off-the-shelf biochemical (and similar) reactions in water

Note also that the choice of diamondoid structures per se was, I think, used in the original writings more because it can be relatively tractably analyzed than because all the interesting structures to be made are just mechanical diamondoid machines. There are of course many interesting material and functional properties, electronic states, and so on, that one could make given a general ability to positionally direct chemistry — but most of these have surely not been thought of yet.
In this aforementioned series of email exchanges featured on Richard Jones’s Soft Machines blog, which are very much worth reading for those seriously interested, Philip Moriarty also drills in hard on the experimental practicality (or lack thereof) of scanning probe based mechanosynthesis schemes, drawing attention to issues like spontaneous reconfiguration of the surfaces and tips being manipulated, as well as the reliability of the reactions, and other issues. This isn’t to rule out something in this space being possible. Indeed, a major theme of the exchanges is the disconnect between Moriarty’s desire for specific schemes to critique in detail, and his interlocutor’s desire to capture a broader design space which could be searched for viable schemes given the right kind of funding and scale of effort. Still, it has been >1.5 decades since that exchange and I’m still not seeing published demonstration experiments, or even published plans detailing all of the experimental steps and contingencies, of the kind Moriarty was asking for. Moriarty himself got at least one related grant.
We can see an updated perspective from Moriarty in his interview with OpenPhil, where he says “In order to do this with SPM, researchers need to know more about, and be able to control and recover, the chemical environment at the end of the microscope tip… Speaking optimistically, non-scalable atom-by-atom construction of small 3D objects (i.e. nanoparticles) using SPM techniques might be possible in about 30 years… APM is probably feasible for a relatively narrow subset of materials, in principle, but there will be very substantial engineering challenges. It may be more likely to work with some materials rather than others. For example, silicon materials seem more promising than diamondoid materials due to the greater ease of preparing large atomically flat regions of low defect density silicon surfaces.”
Molecular 3D printing
So how could one go forward instead, into a realm of interesting positional chemistry, if not using scanning probe microscope based manipulation? Answer: Molecular 3D printers. Again, these would be biomolecular machines, operating in water, that themselves do positional assembly of other chemicals. I helped organize a workshop about that back in 2016, following up on an earlier workshop he had done with a group at the the Department of Energy in 2015, and we summarized the outcome to OpenPhil, a bit breathlessly, as follows

DOE program on Atomically Precise Manufacturing
Shortly after this, the DOE actually created a (small) program on atomically precise manufacturing which included a component on “molecular additive manufacturing”, and is funding at least a couple of small projects that gesture in roughly this direction. The program manager was David Forrest, a “molecular manufacturing” enthusiast since the 1980s, who has decades later made it to the DOE and then retired shortly after the launch of the program, for reasons unknown to me. Forrest had decades earlier been involved in responses to criticisms of the idea atomically precise manufacturing

The best materials related to the DOE program’s original goals are likely still these slides from the first pre-program workshop
and this talk
If you want to know where the DOE program went after David Forrest retired, this talk by the new program officer, from 2021, is worth watching
It explains that the atomically precise manufacturing program has been transitioned to “ultra-precision for energy efficiency”, focusing specifically on microelectronics, and is mostly funding SBIR grants to industry. On its own this is a great set of goals — we do need a lot of work on new semiconductor devices for energy efficient computing — but it is mostly diverged from the molecular 3D printer angle.
The talk does contain a lot of fascinating details on the more exploratory, non-SBIR research that Forrest funded in 2018, however. Here is the set of 6 research oriented projects they funded outside of SBIRs (a total of about $8M to this portfolio), and how DOE is transitioning them next

STM stands for scanning tunneling microscope and SPM stands for scanning probe microscope.
Note the presence of DNA origami, and “molecular lego”, namely spiroligomers, as discussed above and below in this essay. You can see that the Shih/Oxford team is indeed working on DNA origami stepper motors

and Schafmeister’s team is progressing on spiroligomer work towards using them for catalysts. It looks like they are proposing to transition the spiroligomer synthesis processes themselves to DOD applications/funders, while the DNA origami (“the project that my management has the hardest time understanding why it should be part of [DOE advanced manufacturing office]… I think this is exciting… but I need help with justifying this for AMO… via energy efficiency…”) is being relegated to a “big ideas competition”. So I don’t think DOE is going to be funding self-assembled molecular 3D printers in the near future.
In the meantime, Turberfield’s group did publish this paper
https://onlinelibrary.wiley.com/doi/10.1002/smll.202007704
“Strategies for Constructing and Operating DNA Origami Linear Actuators”
which acknowledges the DOE funding.
The MEMS STM platform is U Texas in collaboration with Zyvex. Fascinatingly, the talk mentions that the UCLA project on 3D scanning tunneling microscope tip sculpting

has been acquired by the Canadian Bank Note company! (“The UCLA team was substantially delayed when their industrial partner was acquired by the Canadian Bank Note company… we believe this company is now the core of Canada’s approximately $200M atomically precise manufacturing program… they have ordered dozens of the low temperature STM machines… diamondoid tool… probably the lowest TRL level project in the group…”)
Details on the molecular 3D printing concept
Let’s explain the idea a bit more though.
We can define “molecular additive manufacturing” as something like “Fabrication of macromolecular objects using programmable, externally-directed (e.g., light driven or chemically-clocked) positional control to direct (e.g., by activation or deprotection) the covalent chemical addition of molecular building blocks to specific sites in nanoscale structures.”
A “molecular 3D printer” would itself be a nanoscale device — self-assembled from a limited set of materials we can synthesize, like short DNA strands linked to other chemicals — whose purpose is to demonstrate/perform molecular additive manufacturing.
The idea is, again, that the physical body of the molecular 3D printer is ultimately some kind of self-assembled bio-nano-structure (a self-assembled molecular framework), maybe a complicated 3D DNA origami assembly. This has to support precise motion along three axes to position the print head.
So, there would be trillions of these DNA origami like structures, say, each a device a few tens of nanometers on a side perhaps, floating around in a test tube with water and some salts in it.
The motion would be instructed by external signals. Perhaps exposure to one wavelength of light (imagine shining a big flashlight of sorts into this test tube) would drive the X-axis motor to the right, for example, or combinations of pulses at different wavelengths could instruct different motions. Changing the pH would be another controllable parameter that could cause a molecular change which would drive motion. Indeed, there is at least one paper on actuating DNA origami mechanisms using changes in ion concentration. Or, flowing in different DNA nucleotides, or different DNA strands, depending on the design. See Appendix 2 for a discussion of a couple of relevant molecular motor desiggs. All of this was demonstrated conceptually in the Nobel prize winning molecular machines work by Stoddart et al, but would need to be applied to larger structural frameworks like DNA origami. (In scanning probe systems, electronically driven piezoelectric materials are responsible for the fine motions, but we won’t be able to use that in tiny self-assembled molecular devices, alas.)
Meanwhile, chemists have devised macromolecular bricks — like spiroligomers– that can “click together” in precise alignment, and that could be attached to DNA “handles” that allow them to be attached to and manipulated by DNA origami. These bricks can be made large enough, and the binding sites they might bind to on a workpiece spaced far enough apart, that even a relatively low precision positioner (say ~2 nm resolution) might be able direct them to react with one binding site versus its equivalent neighbors.
In light of these advances, we argued that it is now possible to construct each of the three core components of a molecular 3D printer: a) a rigid 3D framework for structural support, b) three axes of nanoscale programmable motion control, c) a set of nanoscale building blocks and site-specific reactions that allow those building blocks to “snap” into defined binding sites on a printed substrate.
Importantly, molecular 3D printing does not require “atom by atom” manipulation, but rather only site-specific deprotection and site-selective assembly on a substrate, coupled with positional control over which sites are de-protected at each step of printing.
So in summary, we’d be talking about something like this
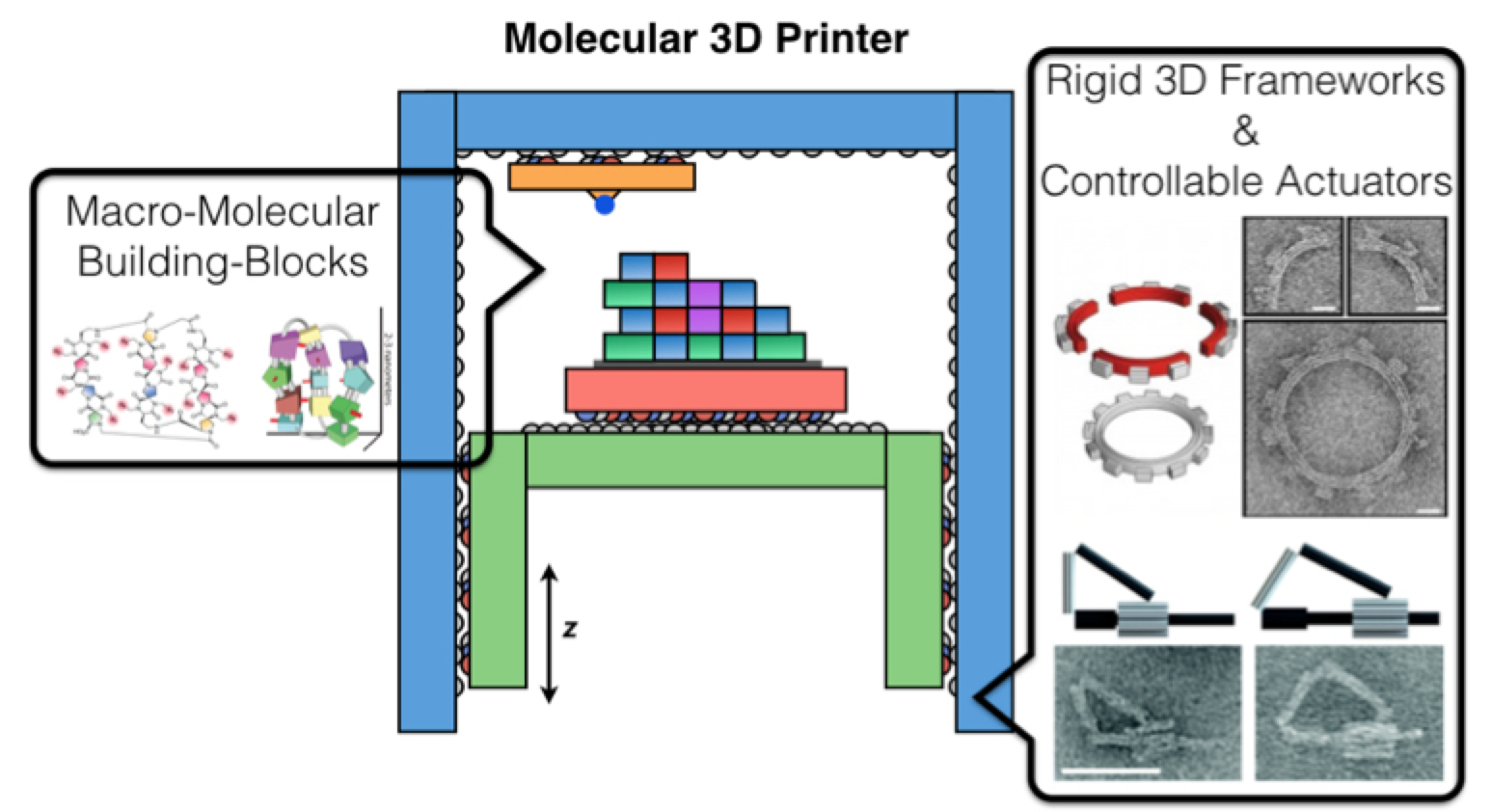
Or a bit more abstractly, putting several components into a system (I am stealing from some Drexler 2016-era slides in the next few images)

which can carry out a sequence of operations like the below, where we can selectively activate sites for bonding, and then deliver new bricks to them just by diffusion in the solution
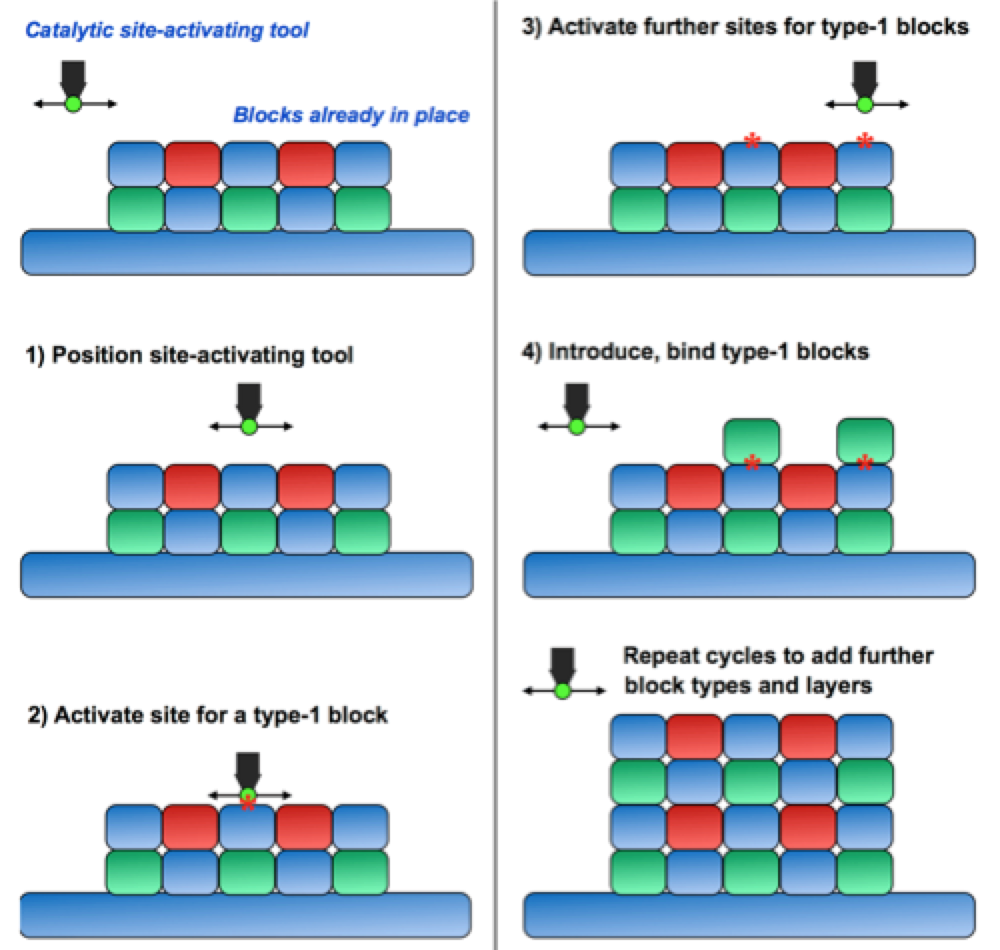
while taking advantage of the fact that we can make the manipulator stiff enough (note for example that a printer made out of diamondoids would be stiffer than a similarly sized one made from proteins, which would be stiffer than one made from DNA scaffolds, generally speaking), and the spacing between potentially equivalent reactive sites comparatively large enough (perhaps ~2 nm spacing), that thermal jiggling doesn’t prevent us from activating just one site without activating its near neighbors, like this
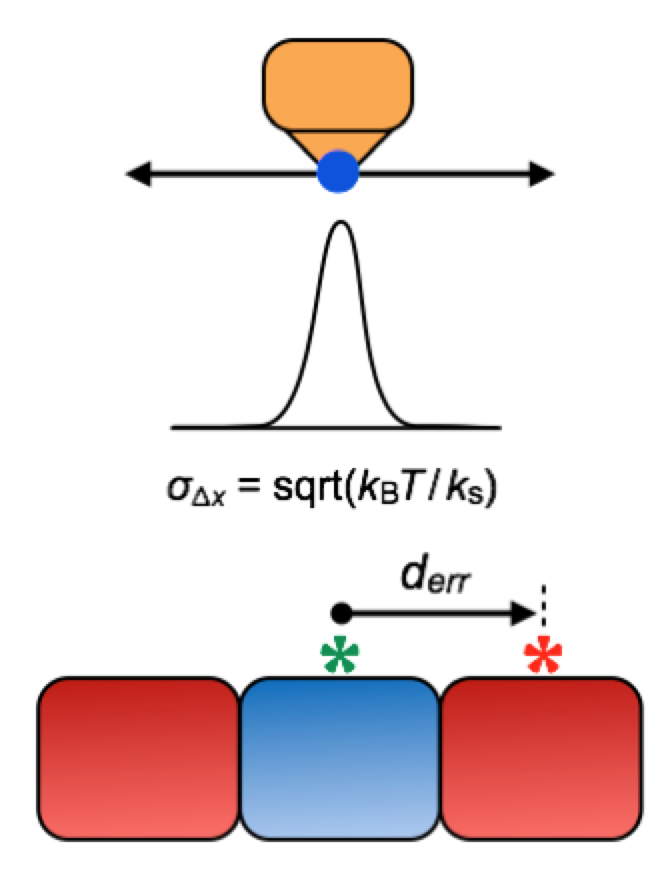
Is that degree of positional accuracy possible with DNA origami? Yes, according to the paper cited in the next image, which claims sub-nanometer control of a distance variable inside a hinge formed from two rigid DNA origami bricks (made rigid by packing many DNA helices closely together):
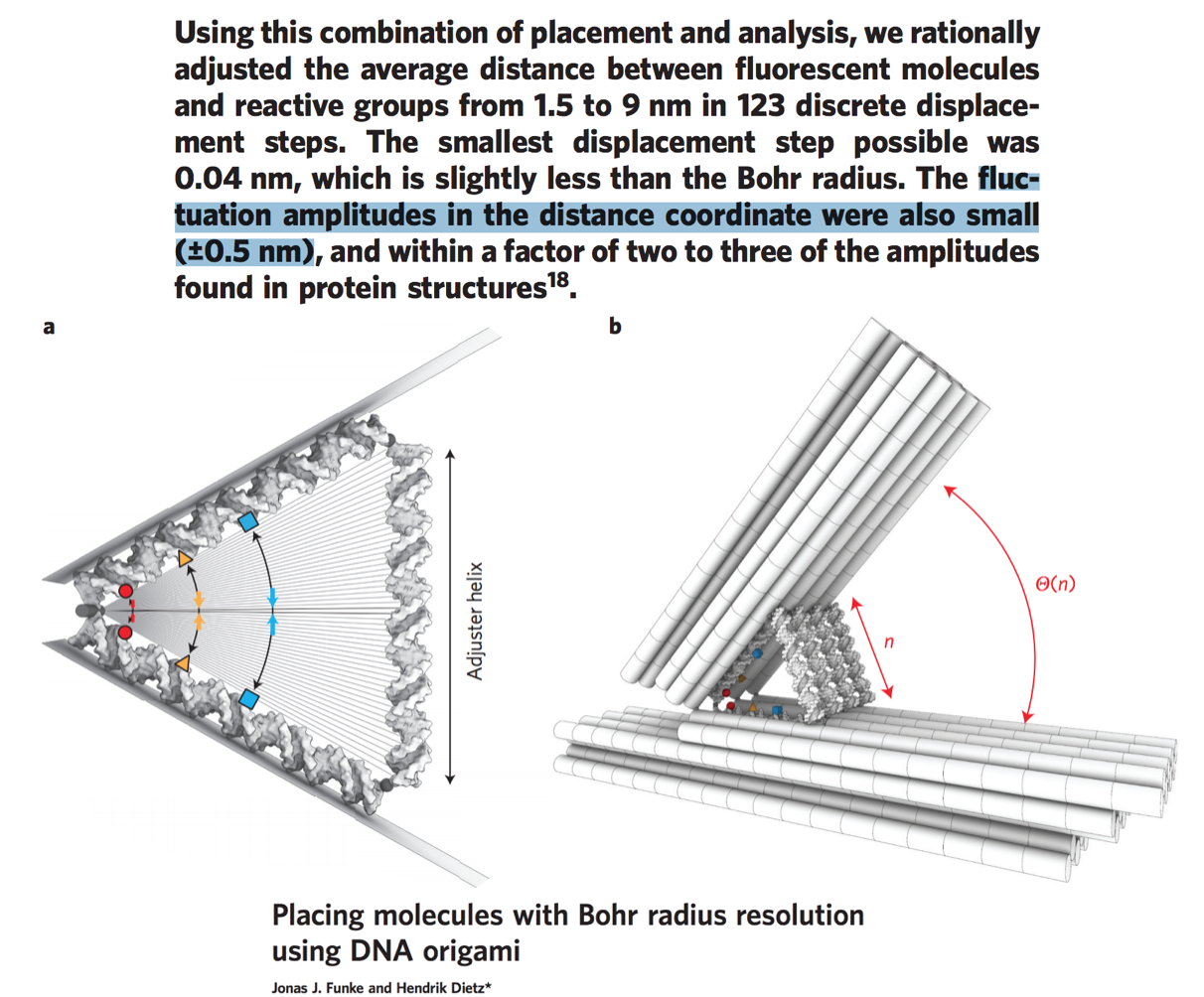
Admittedly, this sketch is vague on a number of elements. It is light on, for example
+ The exact nature of the chemical bonds in the system
+ The exact properties (e.g., electronic, magnetic) of the fabricated systems
But that’s the point of what a DARPA-like program does: it specifies a set of requirements, and a breakdown into subsystems and responsibilities within a project, and otherwise lets the research community creatively respond. Then, winnowing the responses down and establishing some mutually compatible sets of options would be the kind of thing that a DARPA program manager would do with seedlings and early phases of programs. (Galvanizing the realization of prototype full systems would be the kind of thing a full program or series of programs would aim for.)
For molecular 3D printers, you could have requirements on each component like:
Suggested criteria for applicable positioning mechanisms
a) Enable programmable control of displacement (e.g., sequences of discrete steps).
b) Motion is reversible or resettable to enable repeated use.
c) Mechanisms and signals support 2D displacement (already have 1D chain synthesis).
d) Mechanisms provide reliable displacement to enable substantial product yield.
e) Mechanical properties enable adequate constraint of fluctuations.
f) System enables high number density (want macroscopic quantities of product).
Suggested criteria for a functional “printer” structure
a) Structural elements can be combined with stepper motors
b) Potentially enables 2D positioning of an active element
c) Enables a useful range of motion
d) Can adequately constrain thermal fluctuations
Suggested criteria for compatible building-block chemistries (see the book Bioconjugate Techniques for inspiration)
a) Blocks have potentially diverse structures and functionality
b) Can be transported to binding sites by solution interchange
c) Can bind to form stable 2D or 3D structures
d) Can be deprotected/activated at specific sites (e.g., controlled by the printer tip) to direct binding
e) Deprotection/activation site spacing is large compared to mechanical fluctuations of the printer tip
The DOE atomically precise manufacturing program research funding, circa the brief period around 2018 when David Forrest was driving, at least takes some small steps in these directions. Here are some slides from a funded project at Harvard and Oxford on using DNA origami to prototype 2D motion and positioning for a kind of prototype non-covalent molecular 2D printer

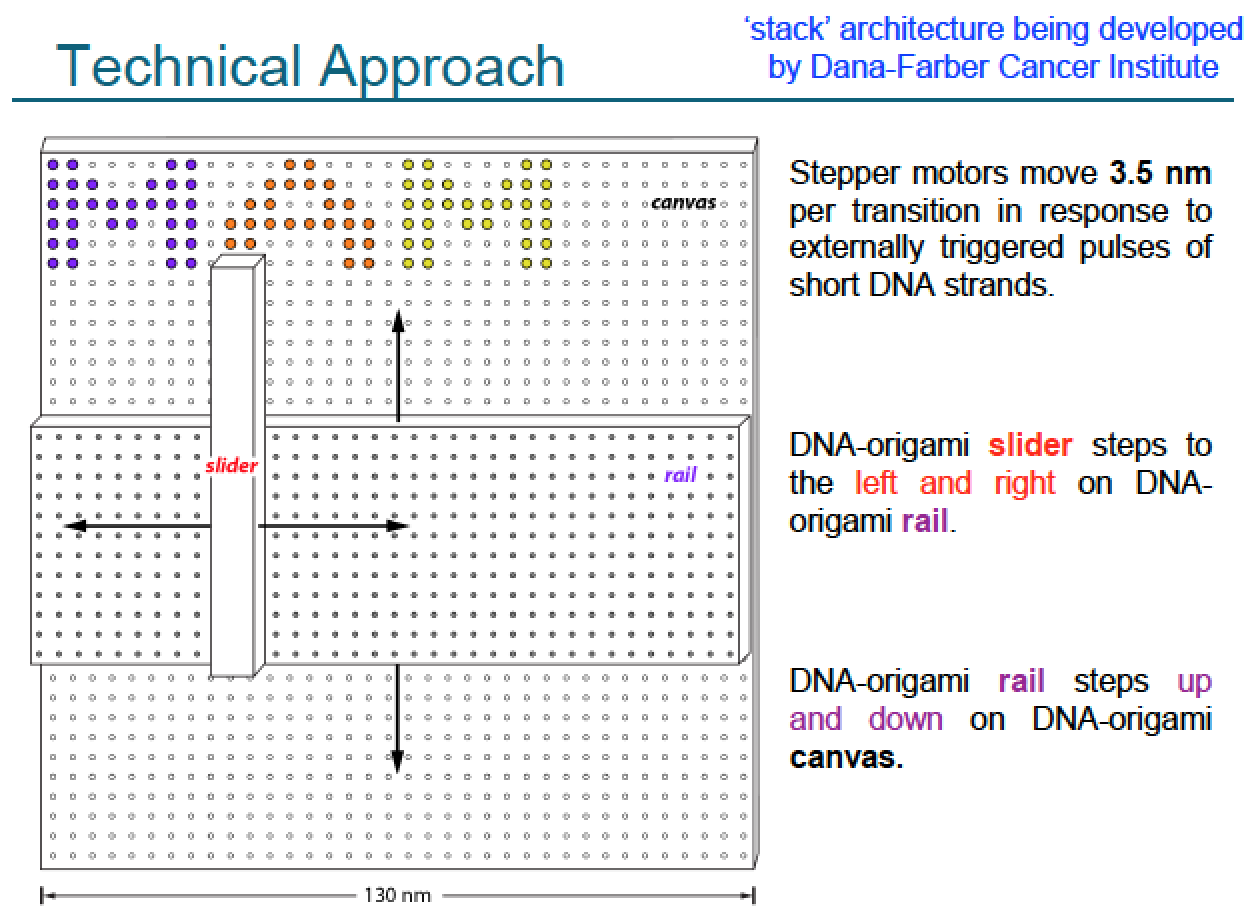
This is great, but just a first baby step towards the full molecular 3D printer goals. Note how in the context of a funding program on molecular 3D printing, the scientific abstracts definitely drink the positional chemistry Kool-Aid— Forrest finally getting to the point of a federal funding program in this area was a big leap forward, albeit apparently short lived.
Previous incarnations of the concept
Looking back through the history, the molecular 3D printing concept or ideas closely related to it have actually surfaced many times:
- In the previously mentioned Moriarty/Phoenix emails from 2005, Phoenix suggests something a bit like the molecular 3D printer idea:

[Moriarty was not impressed, saying “Moreover, and as highlighted in my correspondence with Chris, if we’re carrying out directed assembly of self-assembled blocks then we’re not doing molecular manufacturing in the CRN or Drexler sense.” But I think he somewhat missed the point.]
- The 2007 Battelle roadmap relatedly called for “self-aligning building blocks that enable AP [atomically precise] results from less-than-AP positional control during assembly”.
- In the workshop proceedings it also defines something similar, calling it “numerically controlled molecular epitaxy”.
- We define “numerically controlled molecular epitaxy” (NCME) as the process of depositing atomically precise building blocks—typically molecules—so as to build an atomically precise product. NCME is an intermediate technology, more restricted than fully mature mechanosynthesis, but nonetheless capable of a wide range of useful products, possibly including mechanisms themselves capable of NCME.
- In the workshop proceedings it also defines something similar, calling it “numerically controlled molecular epitaxy”.
Back in the day, I think it was very unclear how the positioning and/or activation/deprotection of such blocks would have worked if not using conventional scanning probe systems, whereas now the ideas of DNA nanostructure based scaffolding or motors, for example, are becoming more palpable, as are ideas of macro-molecular bricks or blocks with multiple distinct activatable or deprotectable functionalizations at different sites.
- There is also a paper from Merkle on a “molecular building blocks” strategy which proposes linking small adamantane-based building blocks in a vaguely molecular-3D-printer-like style, and even some follow-up including a proposal to link adamantane units to DNA.
- There is the part of the 2007 Battelle roadmap document that outright called for, basically, molecular 3d printing (“MMCN” in the below screenshot stands for “modular molecular composite nanosystem”, something like a DNA-scaffolded protein assembly where the proteins can perhaps bind metals or other small bespoke structures):

This was post DNA origami (but pre rigid 3D DNA origami which arose in 2009) and 14 years later we’re in essentially the same position with respect to the project being clearly something one could seriously work on.
- In the workshop proceedings of the 2007 Battelle workshop, Schafmeister proposes a scanning probe based positional 3D assembler of spiroligomer bricks, and similar ideas could potentially be applied in the context of a molecular 3D printer rather than a scanning probe based 3D printer. Note that this one uses direct positioning of the bricks rather than just site-specific activation/deprotection.

- Here is a zoom on some details of that

- Drexler also (unsurprisingly) proposes something similar to a “molecular 3D printer”, in an appendix of the book Radical Abundance (2013)


- The OpenPhil report quotes him: “Dr. Drexler has a concept for a self-assembling, biomolecular, nano-resolution 3D printer operating in solution. The active head of this device (analogous to a printhead) could be moved by linear stepper motors with displacement increments of about a nanometer, operating along three axes and controlled by external optical inputs. Such a device could also be accurate to a resolution of a nanometer (though the system’s components would not be stiff enough to enable accurate positioning at the small-molecule length scale, due to thermal fluctuations), and would construct objects out of biomolecular materials. One way the active head of a 3D printer could work is by removing protective groups from active sites on a surface, allowing the feedstock materials in the solution to bind to the selected sites, transported by Brownian motion. The active head would not “pick up and place” molecules (another common misinterpretation of Dr. Drexler’s ideas that became a target for many critics).”
Where we stand on molecular 3D printing
So where does this leave us on molecular 3D printing? Somewhere like here:
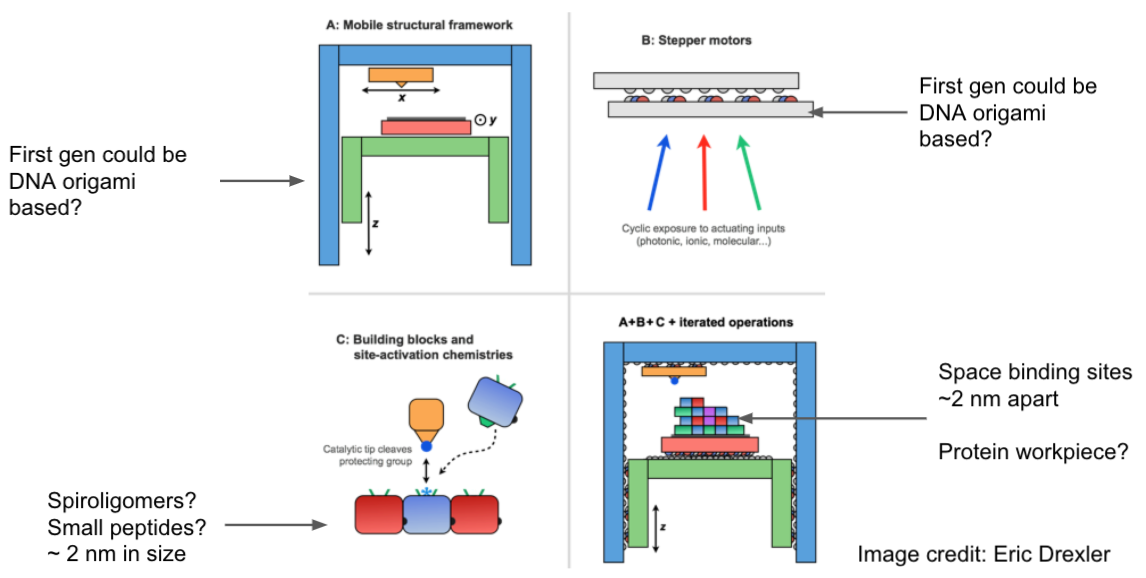
There are probably many options. Here’s a quick sketch at a table putting the ones we’ve focused on in context
| DNA origami based molecular 3D printer of spiroligomers | Scheme where the tip only does catalytic deprotection or activation of sites | … | |
| Scaffold | DNA origami | DNA origami | … |
| Stepper motor | DNA origami based, actuated by with light via azobenzenes or with DNA strand displacement | DNA origami based, actuated by with light via azobenzenes or with DNA strand displacement | … |
| Bricks | Spiroligomers, linked to DNA staple on origami positioner tip | TBD | … |
| Brick size | ~2-3 nm | TBD | |
| Delivery of bricks | Brought to correct position by DNA origami print head, to which it is attached via DNA staple | Diffusion in solution | … |
| Position-selective attachment | Effective concentration enhancement of spiroligomer brick joining | Site specific catalytic deprotection on the workpiece | … |
| Detachment of brick from tip after attachment to workpiece | DNA strand displacement and/or photocleavable bonds | NA: tip only does site specific catalytic deprotection | … |
| Base layer of workpiece | Peptide lattice displaying uniform base layer of spiroligomer bricks? | TBD | … |
| … | … | … | … |
In addition to fleshing out such a design(s) and then doing a ton of work on its pieces, a research program would want to ask questions like: can a printer that is structurally made from DNA origami eventually make a printer that is structurally made from peptide bricks or spiroligomers? Can that printer make a printer that is made from something even better (e.g., stiffer), like some kind of metal-organic complex? And so on, eventually getting to, say, a diamondoid printer. Even in theory. This could be a modeling exercise.
In my view, this is all actually appreciable progress from early sketches of “pathways” forward in positional chemistry from Drexler’s 1992 Nanosystems book, like this

How might we accelerate directed progress?
One could organize a series of ARPA–style medium-scale programs. Here are some ideas for ARPA-like projects or programs:
- Actually doing experiments on the Drexler/Foster proposal from the early 1990s
- Molecular 3D printing
- Extending the ideas of the DOE program, e.g., the Shih/Turberfield project, to a larger scale and more tightly coordinated project
- With multiple phases of developing components, integrating components into systems, and iterating on them
- Again, could use something like a 3D DNA origami scaffold and motors, and something like spiroligomers as bricks
- Needs details on “off the shelf” activation or deprotection chemistries that are appropriate for a given system
- “Protein carpentry” development
- Leveraging machine learning tools, like AlphaFold, and other improvements in computational protein design
- Perhaps aiming for a DNA-origami-like fully addressable surface, but made of protein instead of DNA
- This would be a workpiece for a 3D printer, and a step to creating 3D printers out of (stiffer, finer-grained and more chemically controllable) protein scaffolds rather than DNA origami scaffolds
- Alexis Courbet, in David Baker’s lab, also has the beginnings of a working, designed, protein rotary motor
- Facile design and fabrication of spiroligomer based molecules as “bricks” for assembly
- Building infrastructure for these to be used
- See ThirdLaw Technologies, a startup going in this direction
- Building on ACTION-PAINT as a primitive form of molecular printing
- Pushing something like what CBN Nanotechnologies is doing, in the USA or elsewhere beyond Canada
- An open source “Nanosystems 2.0 with modern computation”
- Open source simulations of long-range and medium range systems
- Simulations of mechanically controlled chemistry
- Note: Computers are much faster now than in Drexler’s day
- Molecular mechanics context in which you can describe reactive chemistry, using ML derived potentials…
- Accelerated DFT calculations
- Made web-accessible and open source so chemists can verify and tweak
- Such a computational study could work backwards toward implementable reaction schemes for positionally directed chemistry
- Can it simulate printers that can make printers, back to something we might be able to make with something we might be able to make starting with a DNA origami based 3D printer?
- Constraint based design languages for nanotechnology
- One of the major problems we see, in discussion around this whole area, is that people like Drexler think in abstract terms about systems, whereas chemists tend to think more about specific molecules. Can programming language design of some kind help to specify constraints which can then be “compiled down” onto possible molecules? Something like: “make me a molecule with 8 carboxyl groups where they are oriented radially in a mechanically rigid way and their tips are no closer than 1 nm apart”, “add a radical out of the plane of the carboxyls and sterically protect it in the absence of light activation”
- Perhaps inspired by programming based CAD software
- Scafmeister is going roughly in this direction with his CANDO software:
- Several other topics identified in the 2007 Battelle roadmap
- Other possible alternative paths, e.g., successive miniaturization of MEMS, although I think this seems unlikely to work
- Other, perhaps ancillary ideas, like “molecular machines inside metal organic frameworks”, towards applications of the synthetic chemical / supramolecular chemistry approach to molecular machines (see Appendices)
Commercial obstacles and institutional needs
Even with such aggressive, directed programs, nanotechnology would still face major challenges. One key problem is that from an economic/commercial front, general purpose fabrication technologies can take a long time to get up and running — for any given single product you already know you want to make, there is likely some bespoke way of making that already, and it will be a while before the general purpose technology can compete on its own turf with any given such special purpose fabrication method. As a result, startups in this area will generally have a very high probability of pivoting.
Take, for example, mRNA vaccines, which come packaged in little lipid nanoparticles. Presumably with downstream development of DNA origami type approaches, not to mention advanced positional assembly, you could make something in that category, but even more powerful, tunable and generalizable, and indeed atomically precise and perhaps with interesting stimulus-response logic built in. But for now, there is enough work to do “just” to get a good manufacturing process for the specific lipid nanoparticles used to deliver mRNA, and thinking about that as an instance of a “general purpose” fabrication method, rather than just a goal to take the shortest path to, would be a huge distraction. Same for conventional computer chips — trillions of dollars have already been invested in optimizing lithography, so any generic scanning probe or self-assembly or other new general purpose nanotech based assembly method faces an uphill battle to get anywhere near that performance level, for applications people care about.
This may be one reason that “novel materials properties at the nanoscale” are easier for researchers to focus on and still pay the bills, compared with general purpose nano-assembly methods. Quantum computing has done well moving into the commercial space despite all this, in part because of some very unique applications relative to government/security, and in part because of its very crisp theoretical basis (and inherent theoretical interest) and legitimacy within physics.
Overall, this is probably a good example of an area that needs “system building” work, of the type Ben Reinhardt describes in his essay on the need for a new private ARPA-like entity.
Further reading and watching
- Productive Nanosystems A Technology Roadmap
- Unbounding the Future: Chapter 5
- DNA Origami: Folded DNA as a Building Material for Molecular Devices (video)
- There’s plenty of room at the bottom
- Book review: the machinery of life (video)
- Molecular additive manufacturing (video)
- Protein-based assemblies and molecular machines (video)
- Build sequences for mechanosynthesis (patent)
- Molecular 3D printer parts and systems brainstorm (video)
Acknowledgements
Thanks to Eric Drexler for discussion over many years and for some specific images from slides, Ben Reinhardt for some of the high level orientations and ideas particularly around what Private ARPA (PARPA) programs could do here as well as for pushes to consider more concrete goals and alternative paths, Michael Nielsen for thoughtful critique around purpose and framing, Ales Flidr for galvanizing many early discussions, Ben Snodin for his relevant analysis work, Daniel Oran, Ashwin Gopinath, George Church and Robert Barish for helpful nanotechnology discussions over the years, David Forest for establishing the relevant DOE program and initial workshops, William Shih, Shawn Douglas, George Church, Ralf Jungmann and Peng Yin for mentorship in the nanotechnology field some years ago, and the participants and organizers of the 2016 workshop on molecular additive manufacturing held at Cambridge University, particularly Andrew Turberfield, William Shih and Christian Schafmeister for willingness to dive into details at that workshop. Thanks to Jose Luis Ricon for general suggestions and Geoff Anders for a close read of the and suggestions of key clarifications of the argument.
With apologies for much important work that I didn’t have time or awareness to cover here.
Appendix: What are covalent bonds anyway?
Before letting nanotechnology seem too abstract, it is good to go back to basic physical organic chemistry, and understand what covalent chemical bonds really are. Here are some good links for that from The One Background Class You Need To Really Understand The Content of This Essay, namely James McBride’s Freshman Organic Chemistry at Yale. I recommend watching the whole course end to end if you have serious interest in nanotechnology or related — it covers everything from the quantum mechanics of bonding, to how molecular simulations work, to organic functional groups and reactions, scanning probe methods, the structure of peptide chains… basically all the fundamentals needed to understand the above material at a more fine-grained level.
Here are some parts that deal specifically with the fundamentals of bonding:
http://chem125-oyc.webspace.yale.edu/125/quantum/end/end.html
http://chem125-oyc.webspace.yale.edu/125/quantum/tunnelbond/tunnelbond.html
Here are two other nice links showing the quantum mechanics of what a covalent bond is:
http://cs.westminstercollege.edu/~ccline/courses/phys301/Knight/Knight_41-9.pdf
Anyway, here is the key picture to understand for that — a low energy bonding orbital and a higher energy antibonding orbital arise even in a one-dimensional double-well potential analog of chemical bonding, if the spatial overlap and energy match between the parent orbitals of that bond are strong:
Amazingly, this connection was figured out in just the half decade or so from the publication of Schrodinger’s 1926 paper on quantum wave equations, taking advantage of a lot of chemical lore that had been figured out more empirically before that. See the writings of Linus Pauling.
A major reason to include this is that I want to suggest that the chemistry and physics underlying the fabrication methods proposed, even in the most far out areas of nanotechnology like positional chemistry, are not that mysterious at their core. You could certainly make structures with novel materials properties using these methods, things we haven’t thought of, and there is a very interesting design space that opens up, but the principles underlying the fabrication processes themselves are fairly basic.
McBride’s course explains, for example, how in many cases, to understand chemical reactions, you just need to think about the overlap and energy match between a highest occupied molecular orbital (HOMO) on one molecule, and a lowest unoccupied molecular orbital (LUMO) on another, and then to think about breaking down those orbitals into bonding and antibonding orbitals formed from those of the constituent atoms.
That gives a very “designable” understanding of reactivity, including of the 3D spatial aspects like what angle a group would have to come in at to kick off another. This is amenable to DFT and other standard ab initio chemical simulation methods.
Consider for example this paper on “Mechanically controlled molecular orbital alignment in single molecule junctions“, which uses molecular orbital concepts together with mechanical force concepts to explain conductance in a molecular electronic device: “ …by applying a mechanical force to a molecule bridged between two electrodes, a device known as a molecular junction, it is possible to exploit the interplay between the electrical and mechanical properties of the molecule to control charge transport through the junction… Counterintuitively, the conductance increases by more than an order of magnitude during stretching, and then decreases again as the junction is compressed… we attribute this finding to a strain-induced shift of the highest occupied molecular orbital towards the Fermi level of the electrodes, leading to a resonant enhancement of the conductance.” We can understand this stuff pretty well, in other words. Likewise with this paper on scanning probe methods from Moriarty, which uses molecular orbitals as a key framework.
Even more so, molecular mechanics and molecular dynamics models, as used by biochemists, say, are basically a picture of bonds as springs + torsional springs, plus force fields for non-covalent interactions. Which is also how people intuitively think of, say, hydrogen bonds and hydrophobic interactions to understand the structure of DNA or protein secondary structure motifs.
Together, these frameworks provide a viable approach to not just simulate, say, protein folding, but also to understand what happens chemically in active sites of enzymes, in not all, but many, cases. The same basic intellectual toolkit that a physical organic chemist or a biochemist uses can be applied to these nanofabrication methods. It is computationally intensive to simulate this stuff, but not fundamentally mysterious.
Appendix: Molecular stepper motor concepts
Here are two more detailed examples, at different levels of abstraction and development, for the externally-controllable 1D actuator concept, as would be used in molecular 3D printers
Actuator Sketch #1: Ratcheting a polymerase forward and backward along DNA
Double-stranded DNA is rigid on the 50 nm length scale, and a polymerase stepping base to base would make discrete 0.34 nm steps. Can this process be controlled bi-directionally in a discrete fashion?
One option would be to exploit a DNA polymerase as the stepping motor.
Suppose we had a repeating DNA template like
3’-ATCGATCGATCGATCG-5’
Let the position of the polymerase be marked as P.
Suppose we have an initial configuration like
P
T
3’-ATCGATCGATCGATCG-5’
And that we only add the letter “A” into the solution. Then P can step forward one letter (since A is complementary to the next base, T), but can go no further, since there is no G.
P
T
3’-ATCGATCGATCGATCG-5’
- A
→
P
TA
3’-ATCGATCGATCGATCG-5’
If we now remove A and add G, we can proceed another step.
P
TAG
3’-ATCGATCGATCGATCG-5’
And so on. This is great, but it only allows you to step forward.
If, however, one uses a polymerase like T4 DNAP which has a 3’ to 5’ exonuclease activity, then we can control the movement in both directions. For instance, if we have all three letters T, A and G in the solution, then P would remain in the same configuration, alternating between chewing back and moving forward, but never able to progress past the present position.
If we now remove G from solution, but keep T and A, we can recess back by one letter due to the exonuclease activity chewing back the G, which then leaves into bulk solution, without another G to replace it.
Likewise, if add C while removing T (e.g., by first removing T and then adding C via two successive buffer exchanges), we can progress one step forward from here, without allowing the polymerase to move forward indefinitely on the repetitive template pattern.
P
TAGG
3’-ATCGATCGATCGATCG-5’
(removed T from solution, so can go no further)
By having a proofreading DNA polymerase as well as a proofreading RNA polymerase, you could have two independent axes of control, each with < 0.5 nm step size, with the 8 DNA and RNA nucleotides as control molecules. Parallelism (e.g., motors in different locations on a surface being controlled differently) could be achieved through microfluidic buffer exchanges or local photo-uncaging of the nucleotides. If the real-time position could be monitored via, e.g., FRET (as used in many single molecule biophysics experiments), then, since the steps can be controlled bi-directionally, it should be possible to detect and correct errors, e.g., a failure to step.
Actuator Sketch #2: High-level design principles for a 3-phase stepper motor
The second sketch is an explanation of this patent, and was used in the intro materials to our molecular 3D printing workshop in 2016.
Stepper motors are a class of linear actuators that can controllably progress one step at a time, forwards or backwards, and then remain rigidly in position. The 3-phase scheme below is basically a translation into the molecular domain of the most common principle used in macroscopic linear stepper motors:
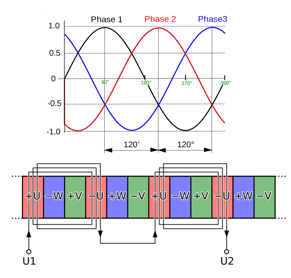
They work by leveraging multiple kinds of interactions, which can be switched on and off individually. This kind of idea can be implemented at the molecular scale just as well as at the macroscale.
A periodic patterning could enforce a periodic background interaction potential between two solid nanoscale objects, like 3D DNA origami. Two other, switchable, periodic patternings are shifted relative to the first. When these switchable patternings are turned on, they adjust overall interaction potential. By turning on and off these switchable patternings in particular orders, the equilibrium offset between the two structures can be stepped forwards or backwards in discrete steps.
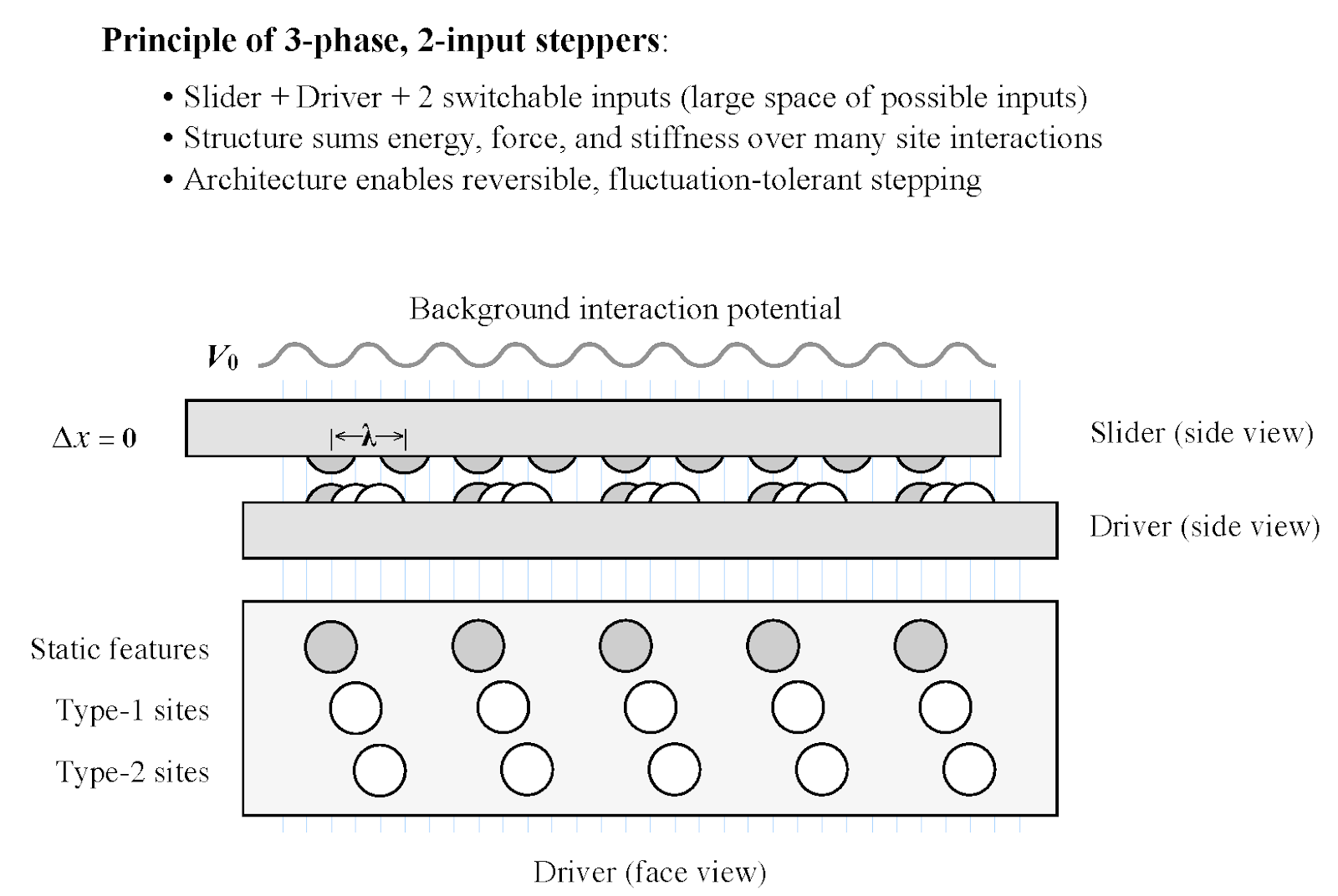
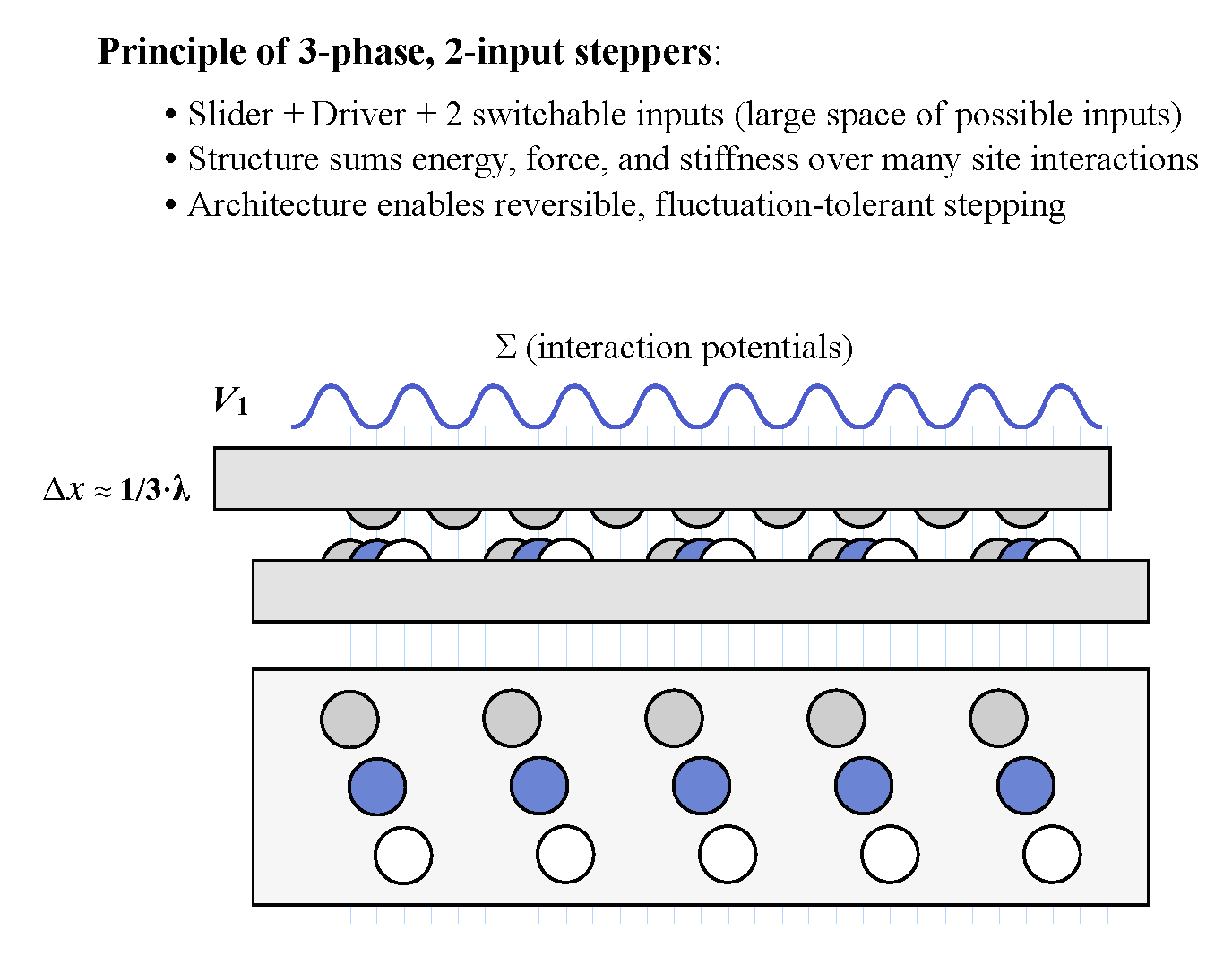
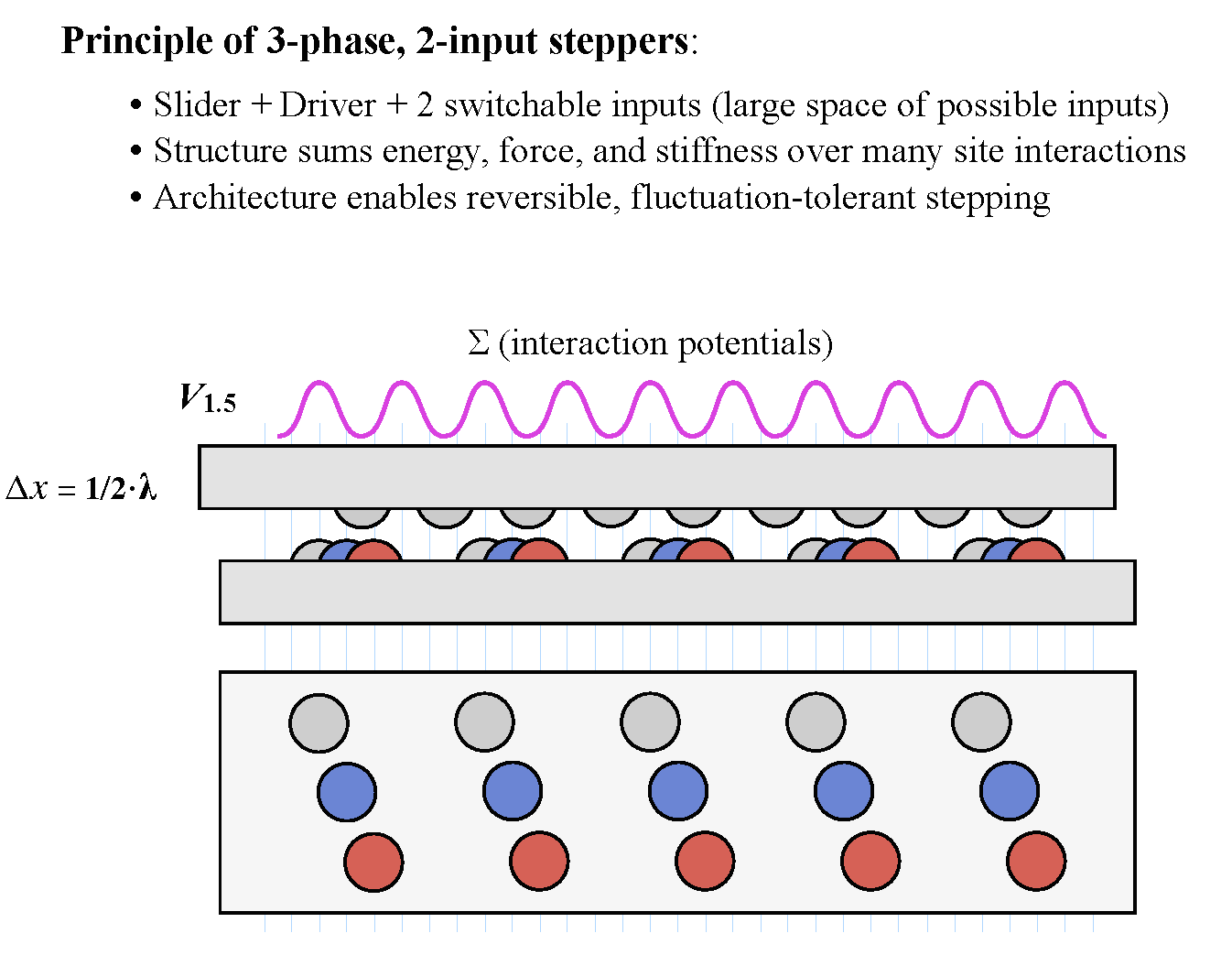
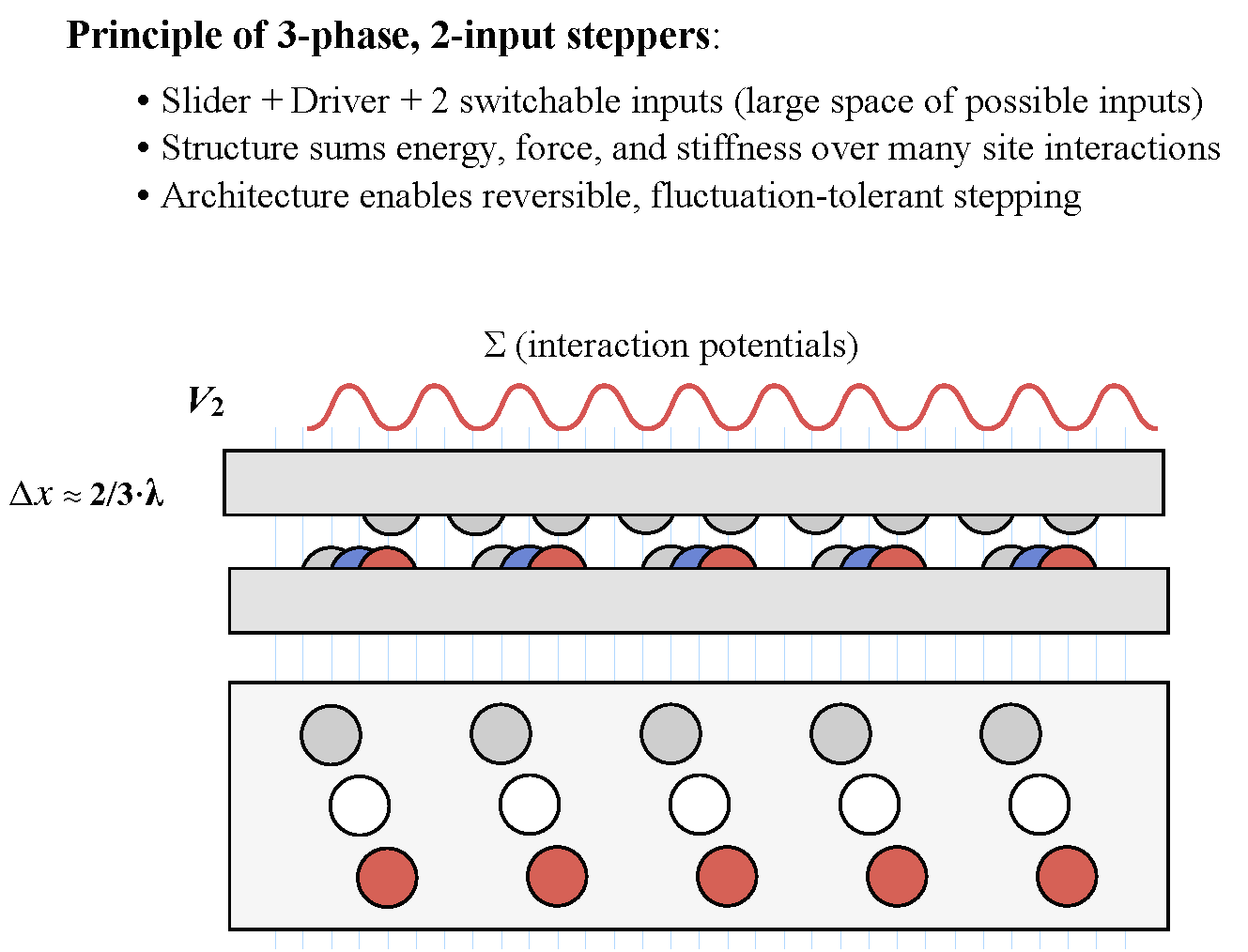
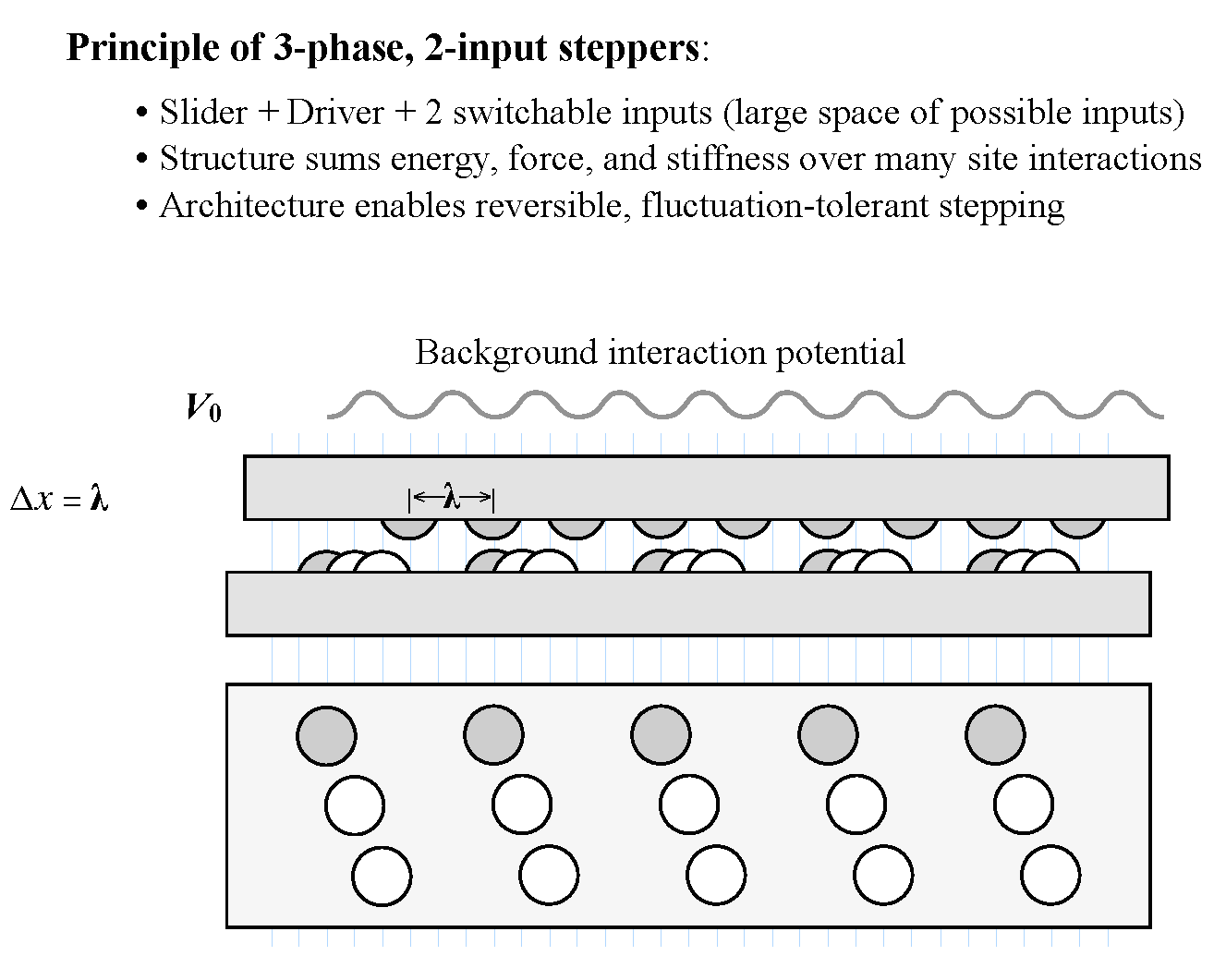
To give an example of how this kind of stepper scheme could be made more concrete:
- Two orthogonal switchable interactions could (through some mechanism) exploit protonation and magnesium binding (each to some appropriate kind of site), clocked by solution interchange; a third kind of interaction (giving three pairwise-orthogonal kinds of site activation and three independent steppers) could use selective calcium binding.
- As for slider and driver structures (made of something or other), the geometric constraints are weak, and the design of an interface that would hold the structures together while permitting translational motion is an interesting question.
- The large number of interaction sites per motor interface is what ensures that adequate mechanical stiffness and tolerance of binding fluctuations, thermal fluctuations, and defects can be engineered.
Appendix: Combining top down and bottom up
Separate from “positional chemistry”, take a look at this passage from one of the early-1990s papers, emphasizing the importance of combining “top down” (like macroscopic mechanical devices such as AFM) and “bottom up” (like self-assembly of molecules on surfaces and using antibodies to grab small molecules), contrasting this approach of positional directing a site-specific assembly of complex molecules, with the IBM work on single atom manipulation

How are we doing on bridging top-down/bottom-up modes of fabrication, to create macroscopic systems with complex aperiodic organization down to the nanoscale?
Explicit work on bridging top down and bottom up
From the same 2006 NAS report: “A bit further out, some scientists envision the use of increasingly sophisticated self-assembly processes, including biomolecular processes… to routinely bridge between the molecular scale and the larger structures that are readily fabricated with the aid of lithography. The result would be the ability to build structures approaching or exceeding biological levels of complexity—a capability that would have enormous implications for information technology, medicine, and energy production, and for endeavors not yet imagined.”
What about work in this area? In my mind, the key goal would be to take the full addressability within DNA origami — the fact that each staple strand, which goes to a unique site on the origami, with few nanometer precision, say, and can thus bring a unique attached chemical, nanoparticle or so on to that particular site on the origami (the 2007 Battelle roadmap has a good description of this concept if you want more, they call it “unique addressing”) — and extend that to an area approaching the size of a computer chip, so say a millimeter on a side instead of 100 nm on a side.
DARPA had a program on this general area called Atoms to Product
https://www.darpa.mil/work-with-us/a2p-performers
but it didn’t focus on DNA origami or using DNA sequences to program assembly.
Perhaps the most interesting experimental demo so far is from Ashwin Gopinath and Paul Rothemund. What they can do is use electron beam lithography to make small “sticky” spots (I’m glossing over the chemistry obviously) on a silicon surface — and importantly, those spots can have a well defined orientation and be of the exact right size and shape to stick to a shape-matched DNA origami. Like this: note how the DNA origami triangles line up quite well inside the lithographic triangles:
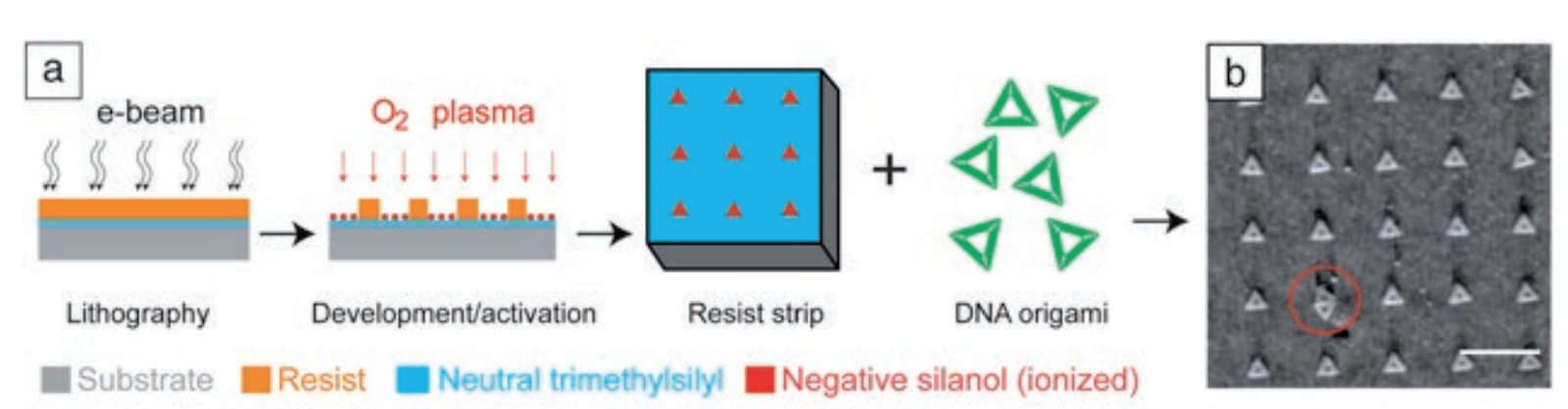
They can then use this to make some cool photonic devices. This is one of those technologies where it feels like it now needs exploration to find its killer app. One possibility is positioning of a small number of discrete photonic components at the right locations on chips, e.g., for single photon sources — there is some progress in that general direction: “the authors were able to position and orient a molecular dipole within the resonant mode of an optical cavity”.
How we might accelerate progress
One idea I like is to go beyond just matching the shapes of lithographic spots to the shapes of DNA origami, and instead to actually have unique DNA sequences at unique spots that could uniquely bind to a given DNA origami. This would be a combination of a few technologies
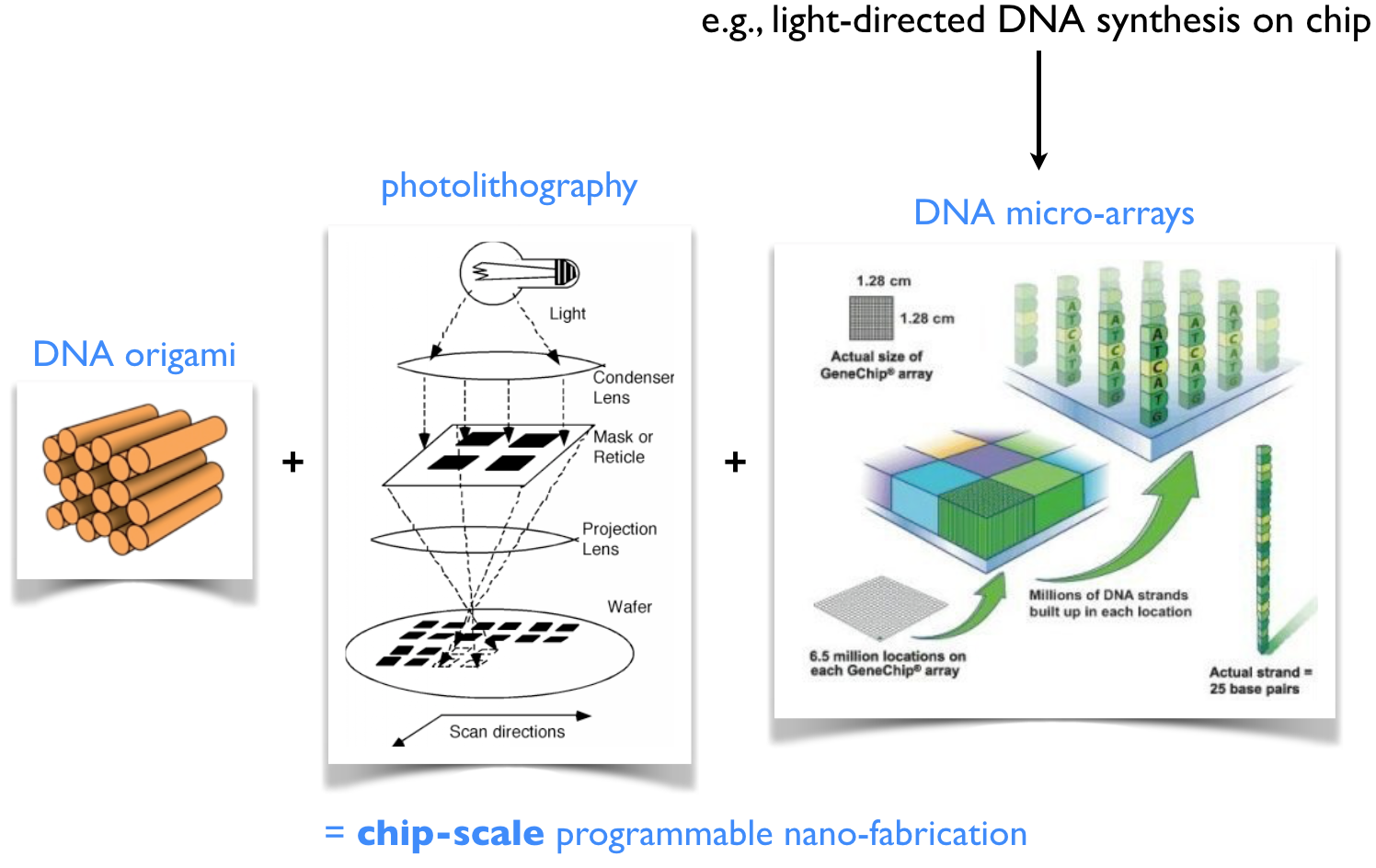
In more detail, in my thesis chapter on “nm2cm” fabrication
http://web.mit.edu/amarbles/www/docs/Marblestone_nm2cm_thesis_excerpt.pdf
http://web.mit.edu/amarbles/www/docs/Bigger-NanoBots-Marblestone.pdf
https://dash.harvard.edu/handle/1/12274513 (for the full references and such)
we proposed that the key gap in this field — of integrating biomolecular self-assembly with top-down nanofabrication to construct chip-scale systems — is that, as impressive are works like Ashwin Gopinath’s using shape to direct DNA origami to particular spots on chips, it would be even more powerful if we could direct specific origami to specific spots on chip in a sequence-specific way: each spot on the chip should have a unique DNA address that could match to a unique DNA origami slated to land there. How can we do that?
1.1) Optical approaches (faster, cheaper than electron beam lithography) can deposit or synthesize particular DNA sequences at particular spots on chip — and this is widely used to create DNA microarrays — but the spot sizes and spacings of the resulting DNA “forests” are too large to achieve “one origami per spot”
http://www.biostat.jhsph.edu/~iruczins/snp/extra/05.08.31/nbt1099_974.pdf
1.2) So we proposed to combine sequence non-specific but higher resolution photolithography to make small spots, with coarser grained optical patterning to define sequences for those spots, and then large origami rods spanning spot to spot to help ratchet orientation and spacing into a global “crystal-like” pattern: see nm2cm chapter above
Anyway, we didn’t demonstrate much of this experimentally at all (alas, it needed an ARPA program not a rather clumsy grad student, or at least that would be my excuse), but since then
1.a) Implosion Fabrication (ImpFab), mentioned above, may now provide a way to take a sequence-specific DNA microarray and shrink it so that the spot size matches achievable sizes of DNA origamis. Something like this: note the tiny DNA origami in the lower right for scale
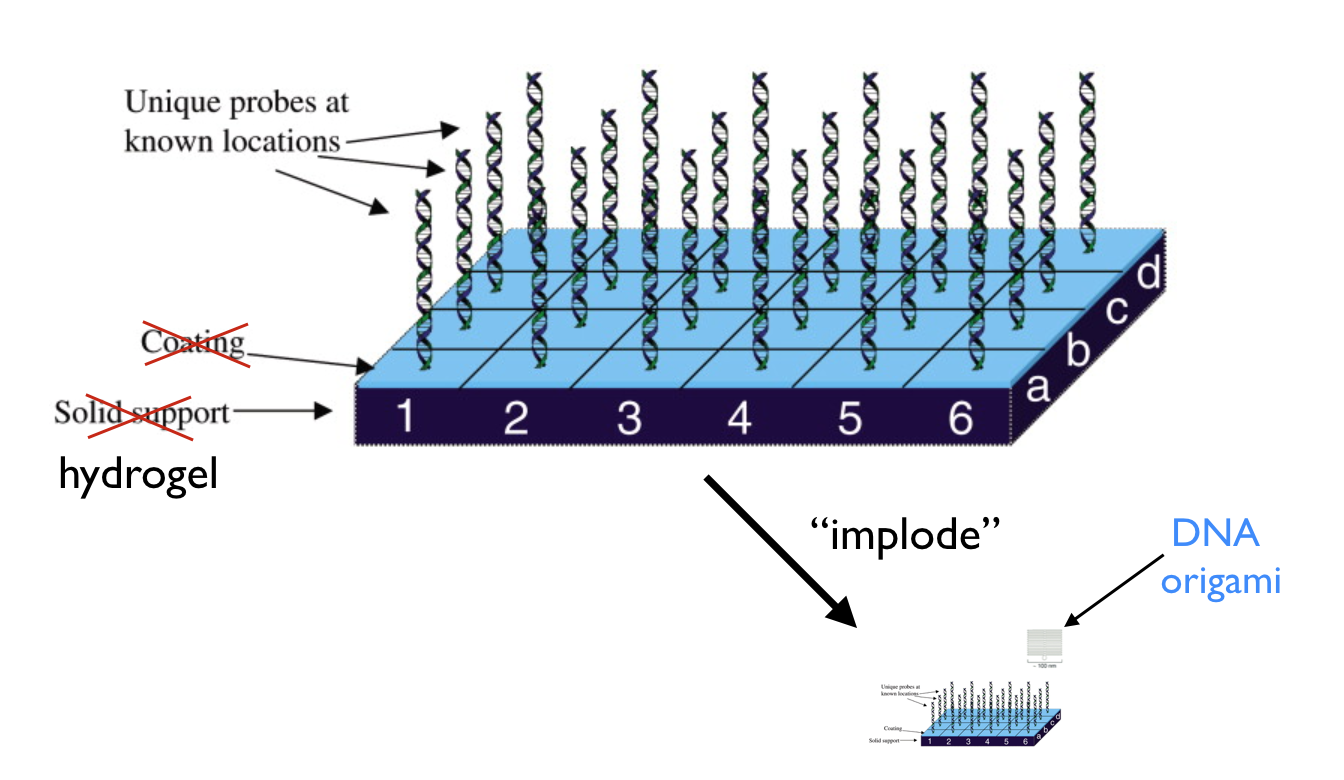
1.b) People have started making smaller/finer-resolution microarray-like sequence-specific (albeit random) patterns on chips, and even transferred them to other substrates
https://www.biorxiv.org/content/10.1101/2021.01.25.427807v1.full
https://www.biorxiv.org/content/10.1101/2021.01.17.427004v1.full
(With these, you make a fine grained but random pattern and then image/sequence it in-situ to back out what is where. This would obviously entail a significant metrology component, i.e., figuring out what sequence is where and then synthesizing a library of adaptor strands to bring the right origami to the right sequences.)
1.c) DNA origami have gotten bigger, too, closer to matching the sizes even of existing non-shrunken microarray spots
https://www.nature.com/articles/nature24655
The proposed approach would achieve sequence-specific addressable chips that can direct unique-sequence DNA origami to specific spots on chip with an exponential diversity of sequence programmability rather than a more limited diversity of shape and surface affinity programmability as in previous work.
To do this, one needs to be able to approximately size-match single DNA origami-like structures with single sequence-specific spots on chip (think of them as localized “forests” of copies of a particular DNA sequence on chip). One way of doing that would be to photo-pattern a sequence-specific DNA microarray, and then shrink the spots with Implosion Fabrication.
Thus, I think sequence-specific bio-chips, in which thousands of distinct origami as defined by sequences, not just a few as defined by shapes, can be directed to their appropriate spots on chip in a multiplexed fashion, should be possible. Exactly what their applications would be is less clear to me.
The lack of usable foundries for producing and packaging emerging nanosystems and of design enablement (e.g., CAD tools, compilers) for designing nanosystems (of a variety of kinds) is also a blocker for progress right now. Because devices are made in a bespoke fashion in the labs of their inventors, rather than mass produced and made usable for end users, researchers who aren’t experts in a given nanofabrication field can’t easily explore potential applications of that field that could drive commercialization. DARPA’s 3DSoC program is trying to help with this for 3D low-energy beyond-silicon chips, but the general need is still there across many other emerging sub-areas of nanotechnology, from DNA origami to implosion fabrication to photonic processors.
Update: IARPA issues RFI on bio-templated microelectronics
Appendix: Other key nano advances since 1990s
Molecular machines via conventional synthetic chemistry
The 2016 Nobel Prize in chemistry was awarded to Stoddart, Sauvage and Ferringa for tiny molecular machines made via conventional organic synthesis methods. These include linear and rotational motors. Often making these involves clever methods for forming topologically locked rings
and then actuating them to move relative to one another using, for example, heat or light
Here’s a more detailed chemical structure of one
These are very tiny (only on the order of 100 atoms), pretty floppy, and don’t do much of anything that’s useful yet — but they demonstrate basic principles for how to create these mechanically/topologically linked structures, actuate them, and achieve unidirectional motion. A lot of the key work on this for which the Nobel was awarded was done in the pre-2005 period.
Synthetic chemical machines used to direct synthesis
In 2013, David Leigh’s group published a paper called “Sequence-Specific Peptide Synthesis by an Artificial Small-Molecule Machine”. A ring structure is threaded around an axle that contains several amino acid groups arranged in sequence. While moving down the axle, successive amino acids are added to a chain grown off of the ring structure. This is vaguely ribosome-like in spirit (albeit the thread is pre-loaded with the sequence of amino acids itself rather than an RNA code that directs which amino acids the ribosome incorporates from solution, and albeit only making very short chains so far).
In 2017, some of the same authors published “Stereodivergent synthesis with a programmable molecular machine“. Nature hailed it as a real step toward a molecular assembler:
“In 1986, the futurist K. Eric Drexler published Engines of Creation: The Coming Era of Nanotechnology1, in which he laid out his vision for the field that became nanotechnology. Engines fired many imaginations, including that of one of the current authors2 (T.R.K.), but the big picture of Drexler’s vision also drew well-founded criticisms3,4 because some of the details were incompatible with real-world constraints. One element of this vision attracted particularly strong censure5: the concept of “molecular assemblers” — nanomachines that “will serve as improved devices for assembling molecular structures”. On page 374, Kassem et al.6 report a non-biological example of what could be regarded as a molecular assembler.”
The paper describes a pH controllable molecular actuator which can shuttle between two positions; during a chemical synthesis it has both an initial and final position, shuttling between, and it interacts with substrate molecules to direct one of four geometrically distinct chemical products to be formed depending on the initial and final shuttle positions.
To call this a molecular assembler certainly seems like a stretch to me — the range of its programmability and spatial control being extremely limited — but it is certainly a step in the general direction of atomically precise molecular machines directing the synthesis of other atomically precise chemicals.
Metal-organic frameworks (MOFs) and machines in MOFs
MOFs are an atomically precise and highly designable way to make periodic crystals with interesting chemical functionality inside the unit cells
You can even exploit the fact that a wide range of linkers can be used to put Stoddart-style organic chemical molecular machines into the MOF unit cell. So this allows such molecular machines to be used in the solid state.
Implosion Fabrication
One of these “obvious in retrospect” ideas. It turns out that there are materials called hydrogels that, when you put them in water, can swell uniformly by a large factor, say 10x along each axis. If you add salt, they uniformly shrink back down. What Implosion Fabrication does is uses a focused spot of light to pattern materials into a swollen hydrogel, and then shrink. So you can get 10x better resolution, say, then the smallest diameter of a focused spot of light, i.e., you can get 10’s of nanometer resolution using light with wavelength of hundreds of nanometers. Alas, not quite down to the level of directing the formation of single covalent bonds. This can be done with a variety of materials, but here is shown just with fluorescent dyes for demonstration purposes

3D beyond-silicon compute
Conventional CMOS computer chips are built by using light to shape metal, semiconductor (different “dopings” of silicon with impurities like boron) and insulator (silicon oxide often) in multi-layer patterns that are quasi-two-dimensional. There has been considerable progress recently in extending this “beyond silicon”, to include discrete molecular components like carbon nanotubes, and to denser, finer-grained interconnects between layers
There is also starting to be some work on DNA-directed assembly of beyond-silicon computing elements such as carbon nanotubes (CNTs). Graphene nanoribbons are another beyond-silicon component that could potentially benefit from incorporating this type of strategy — they are under consideration by, for example, Berkeley’s E3S Center which aims to create revolutionary improvements in the energy efficiency of electronics. From the first two papers, it looks like China is pulling ahead on DNA templated CNTs so the US may now feel a pressure to push on this area.