With Jose Luis Ricon.
Adaptations of some of these notes have now made it into a report here.
See also: our review on in-vivo pooled screening for aging.
Much of the global disease burden arises from age-related diseases. If we could slow or reverse root mechanisms of the aging process itself, to extend healthspan, the benefits would be enormous (this includes infectious disease relevance).
Why now is an exciting moment to take action in the aging field
Early advances (e.g., Keynon et al) discovered genes, conserved in animals across the evolutionary tree, regulating a balance between energy consumption and repair/preservation (e.g., autophagy, mitochondrial maintenance, DNA repair), and drove the field in the direction of metabolic perturbations such as caloric restriction and the biochemical pathways involved. Unfortunately, these pathways are ubiquitously involved in diverse essential functions and there is probably an upper limit to how far these can be safely tweaked without side effects, i.e., we may already be near the “pareto front” for tweaking these aspects of metabolism.
The field has started to pick up pace in recent years with a large gain in legitimacy owed to the formation of Calico Inc, and novel demonstrations of rejuvenation treatments in mammals that go beyond simply tweaking metabolism. Indeed, methods are being developed to target all 9 “Hallmarks of Aging”: Stem cell exhaustion, Cellular senescence, Mitochondrial dysfunction, Altered intracellular communication, Genomic instability, Telomere attrition, Epigenetic alterations, Loss of proteostasis and Deregulated nutrient sensing. Commercially, the field is heating up, with many companies pursuing different hypotheses for interventions towards rejuvenation based healthspan extension.
A good summary of compelling opportunities arising in the past decade is provided by OpenPhil, which includes the following items, for which I here add some details:
- Prevent the accumulation of epigenetic errors (e.g., changes in DNA methylation patterns) associated with aging, or restore more youthful epigenetic states in cells
- Ocampo et al (2016) demonstrated in mice transient, cyclic induction, via a gene therapy, of some of the same cellular reprogramming factors that were used by the seminal induced pluripotent stem cell procedure of Yamanaka et al (2006), giving rise to “partial reprogramming” which appeared to restore cells to a more youthful epigenetic state
- Recent work from Sinclair’s lab (2019) called “recovery of information via epigenetic reprogramming or REVIVER” demonstrated rejuvenation of the retina (a central nervous system tissue) in mice in a manner that appears causally dependent on the DNA demethylases Tet1 and Tet2, suggesting a possible causal role for observed epigenetic changes in aging generally
- This is recently studied somewhat more mechanistically in human cells: “Here we show that transient expression of nuclear reprogramming factors, mediated by expression of mRNAs, promotes a rapid and broad amelioration of cellular aging, including resetting of epigenetic clock, reduction of the inflammatory profile in chondrocytes, and restoration of youthful regenerative response to aged, human muscle stem cells, in each case without abolishing cellular identity.”
- Solve the problem of senescent cell accumulation
- Senolytic drugs (see the work of the Judy Campisi lab, and related startups such as Unity Biotechnology) show a median (as opposed to maximum) lifespan extension in mice on the order of 25%. These are being tested in humans on chronic kidney disease (ClinicalTrials.gov: NCT02848131) and osteoarthritis (ClinicalTrials.gov: NCT03513016).
- Although senescence plays important beneficial roles in wound healing, pregnancy and other functions, and removing some senescent cell populations in adulthood can be harmful, there may be ways to clear or target aspects of the senescence associated secretory phenotype (SASP) without wholesale removal of all senescent cells, and/ or to remove specific subsets of senescent cells transiently, e.g., there is also some basic research work on senomodulators and senostatics as an alternative to senolytics
- Bloodborne factors for reversing stem cell exhaustion and potentially ameliorating diverse aspects of aging
- The addition of youthful bloodborne factors and/or dilution of age-associated bloodborne factors can appear to re-activate aged stem cell populations, leading to increased neurogenesis, improvement of muscle function and many other improved properties
- Potentially relevant old-blood factors include: eotaxin, β2-microglobulin, TGF-beta, interferon, VCAM1 (which may mediate aberrant immune cell crossing of blood brain barrier with age or increased aberrant transmission of inflammatory molecules across the BBB)
- Potentially relevant young-blood factors include: GDF11 (questionable), TIMP2, RANKL (Receptor activator of nuclear factor kappa-B ligand), growth hormone, IGF-1
- Recently, Irina Conboy’s lab at Berkeley published a study in mice showing that simple dilution of old blood plasma via replacement with a mixture of saline and albumin (so-called apheresis) could show rejuvenative effects on multiple tissues. This is an already-FDA-approved procedure. If true it is revolutionary.
From a review of emerging rejuvenation strategies from Anne Brunet’s lab, we have a summary figure:
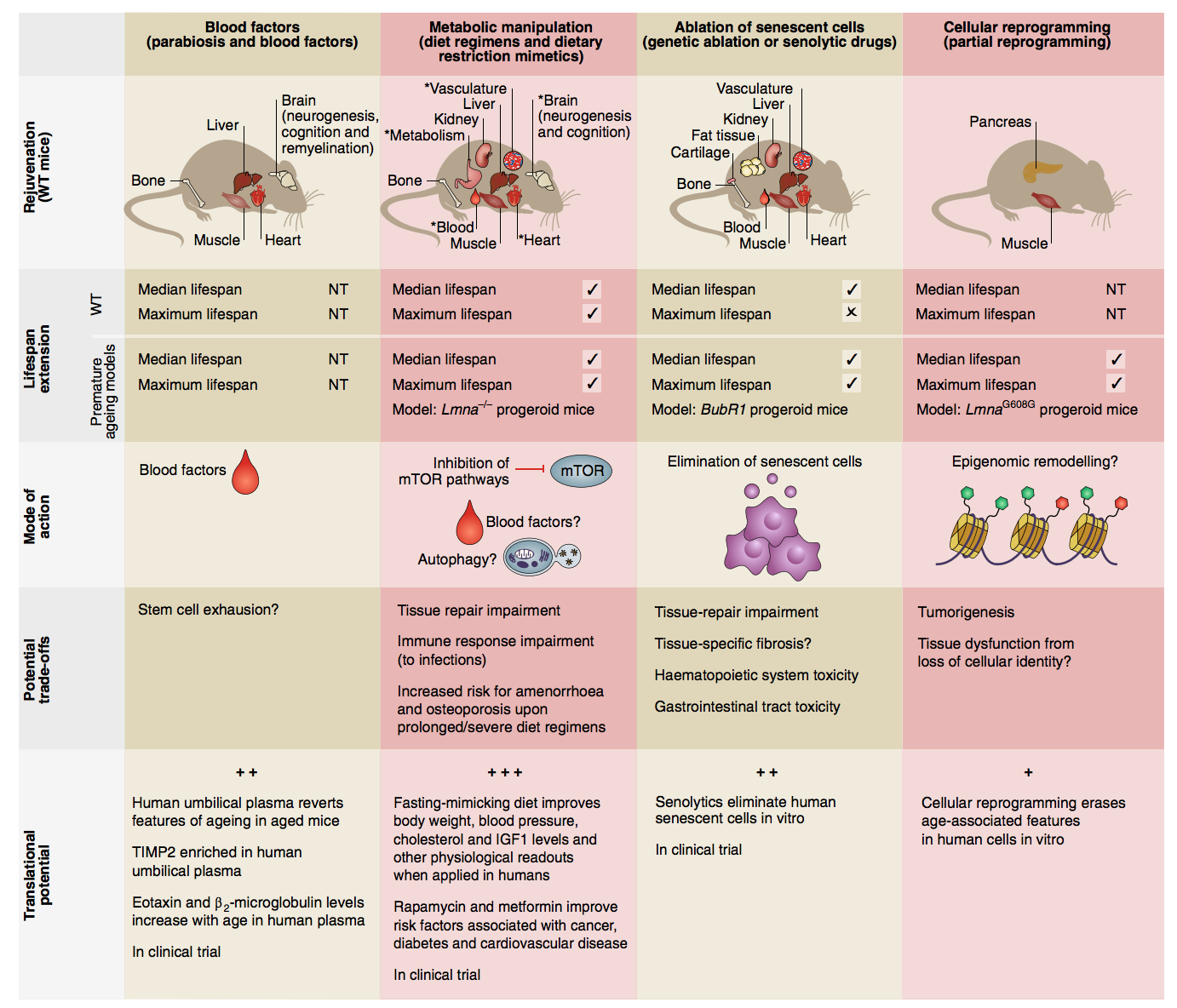
Reproduced from: Mahmoudi S, Xu L, Brunet A. Turning back time with emerging rejuvenation strategies. Nature cell biology. 2019 Jan;21(1):32-43.
In addition, I would add a few other directions:
- Thymic regeneration: the TRIIM (Thymus Regeneration, Immunorestoration, and Insulin Mitigation) study resulted in a variety of beneficial biomarker indicators for epigenetic age reversal and immune function restoration. Greg Fahy gave an excellent talk on this. Combinations of thymus transplants and hematopoietic stem cell transplants can yield profound system-wide effects. (Another company has recently emerged focusing on thymic restoration via FOXN1.)
- Advances in understanding immunosenescence generally, and its coupling to other aspects of aging: e.g., CD38 on the surface of monocytes may be responsible for aberrantly clearing NAD+, which then couples to more traditionally understood metabolic changes in aging — see this excellent summary of the field
- Brain based neuroendocrine control: IKKβ, in the microglial cells in the medial basal hypothalamus in the brain seems to control multiple aspects of aging, and aging genes FOXO1 and DAF-16 seem to be at least in part under the control of neural excitation
- Combinatorial gene therapies: We will discuss the need for this extensively below. In at least one paper, the authors focused on non cell autonomous genes which could drive systemic changes: fibroblast growth factor 21 [FGF21], αKlotho, soluble form of mouse transforming growth factor-β receptor 2 [sTGFβR2], showing they could ameliorate obesity, type II diabetes, heart failure, and renal failure simultaneously (the effect seems mostly due to FGF21 alone)
- Enhancing mitochondrial function: activating the expression of peroxisome proliferator activated receptor gamma coactivator-1α (PGC-1α) and mitochondrial transcription factor A (TFAM) to enhance mitochondrial biogenesis and quality control. T cells deficient in TFAM induce a multi-systemic aging phenotype in mice.
- Novel metabolic targets: e.g., J147, targeting ATP synthase, may not be redundant with mTOR inhibition and thus could be used combinatorially: “J147 reduced cognitive deficits in old SAMP8 mice, while restoring multiple molecular markers associated with human AD, vascular pathology, impaired synaptic function, and inflammation to those approaching the young phenotype”.
“Epigenetic clock” biomarkers, reviewed here, and recent proteomic clocks, are another key advance from the past few years. In theory, clocks like these could serve as surrogate endpoints for trials, greatly accelerating clinical studies, but there are issues to be solved (see below).
Major problems holding back the field today
Still, there are a set of related, self-reinforcing factors that likely dramatically slow progress in the field:
- Studies are often bespoke and low-N, focus on single hypotheses, and only measure a small subset of the phenotype
- I believe that the aging field suffers from fragmented incentives, with many small academic groups competing with one another while needing to differentiate themselves individually — this limits the scope for replication studies, combination studies and systemic convergence.
- In the absence of robust pharmaceutical industry interest in supporting the entire pipeline for anti-aging, including at early stages, the more ambitious studies (e.g., bloodborne factors or blood plasma dilution, epigenetic reprogramming) are being done by academic labs with limited resources, and are therefore carried out by postdocs with incentives focused around academic credit-assignment, i.e., limited incentives for large-scale system-building beyond publishing individual papers.
- For example, the potentially revolutionary recent Conboy lab result that an already-FDA-approved blood plasma dilution procedure ameliorates multi-systemic aging in mice only used N=4 mice for most of its measurements. For such an important-if-true result, this seems absurdly under-powered! Indeed, they apparently replaced the entire first figure of the paper with a post-hoc justification as to why N=4 would give sufficient statistical power, probably in response to critical peer reviewers, instead of simply running more mice. This smells to me like “problematic local academic incentives”, and possibly symptomatic of under-funding as well. See here for more discussion.
- Likewise, the finding of GDF11 as a bloodborne rejuvenative factor has had trouble with replication. That’s the nature of science but it seems prevalent in the aging field.
- Another recent result on bloodborne factors was published with fanfare but had N=6 rats, did not list what their factors actually were, did not list complete methods, and was led by a small unknown company in India — see here for a discussion of some of the problematic aspects in the publication of what otherwise would be a clear win. This study relied heavily on epigenetic clocks.
- The TRIIM (Thymus Regeneration, Immunorestoration, and Insulin Mitigation) study result, which looks preliminarily very compelling, was done with only N=9 human subjects, and was conducted by brilliant but “off-the-beaten path researchers”, working at a small startup (Intervene Immune, Inc) taking donations via its website. This study also relied heavily on epigenetic clocks to argue its anti-aging effect.
- The NIH Interventions Testing program (ITP) at the National Institute on Aging (NIA) specifically exists to replicate and independently test aging drugs, but has apparently not extended beyond studies of single small molecules to my knowledge or addressed the most cutting edge rejuvenation therapies, let alone combinations thereof.
- Tools and tool access/utilization are still limited compared to what they could be
- As in many biological fields, key bottlenecks could be accelerated via targeted engineering advances, but tools companies and tool development projects generally remain under-funded relative to potential impact
- For example, for bloodborne factors, we might benefit from a tool that can very precisely add or remove many specific user-defined factors from circulation, but the field has primarily focused on much simpler interventions, beginning with simply suturing young and old animals together (which raised the confound that the young animal’s organs can filter the old blood, not just provide circulating factors)
- For epigenetic clocks, the technology is still based on methylation arrays, rather than next-generation sequencing based assays which could be multiplexed/pooled to optimize sequencing cost and could then be made much cheaper and thus applied at much larger scale (see below for details)
- For proteomic measurements, these mostly still use defined sets of a few thousand targets, e.g., assayed using SomaLogic aptamer arrays, rather than next-generation unbiased technologies like improved mass spec proteomics (Parag Mallick et al), single cell proteomics (see recent major advances from e.g. Nikolai Slavov’s lab at Northeastern, which are currently operating at very acute sub-scale and have not been industrialized at all), let alone emerging single-molecule protein sequencing (see many new companies in this area such as Quantum–Si and Encodia and recent work from Edward Marcotte’s lab now part of the company Erisyon). Aptamer array based methods may be missing, for example, small peptides like the promising mitochondrial-derived peptide humanin.
- For epigenetic measurements and proteomic measurements, these are mostly not done at single-cell level, limiting our understanding of the specific cell types that contribute causally — for instance, if aging is heavily due to exhaustion of specific adult stem cell populations, we might find that these stem cell populations are the primary locus of the epigenetic changes, but discovering this requires single cell measurement (or else cumbersome cell type specific bulk isolation procedures)
- Epigenetic measurements mostly haven’t taken into account the latest technologies for measuring chromatin accessibility directly, such as ATAC-seq (short for Assay for Transposase-Accessible Chromatin using sequencing) or combined single-cell ATAC-seq and RNAseq
- Measurements of epigenetic effects do not yet include the most advanced in-situ epigenomics measurement technologies that operate by imaging in the context of intact tissues, e.g., FISSEQ, MERFISH, ExSEQ
- As in many biological fields, key bottlenecks could be accelerated via targeted engineering advances, but tools companies and tool development projects generally remain under-funded relative to potential impact
- There is limited infrastructure and incentive for combinatorial studies
- There appears to be a market failure around combinatorial testing of interventions.
- Combinatorial interventions seem warranted for several reasons, including:
- aging may be fundamentally multi-factorial
- there may be synergies between mechanisms that can create self-reinforcing positive feedback loops in regeneration
- using many mechanisms in combination may allow each to be used at lower dosage and thus avoid side effects by avoiding driving any one pathway too strongly
- there may be scientific value in understanding the combinations that turn out to be useful, e.g., for identifying overlapping underlying factors and common effects behind diverse superficially-different sets of interventions
- For the most part, academic labs are not incentivized to systematically pursue combination therapies. This is because they focus on proving out the specific hypotheses that each lab specializes in and depends on for its reputation, often focusing on telling simple stories about mechanisms. For example, while there are labs that specialize in senescent cell clearance or bloodborne factors, it is hard to differentiate oneself academically while having a lab that combines both or is hypothesis agnostic. (Technology/tools focused labs may be better for this but then may lack the long-term follow-through to make the scientific discoveries in this field or to operate large in-vivo studies.)
- It is also simply a lot of work to build up enough expertise in multiple domains to properly combine interventions and measurements, and this may put combination studies beyond the scale of most academic projects, even if well funded, which involve only 1-3 first-author grad students and postdocs playing primary driving roles due to the needs of academic credit assignment.
- Tools in the field are also not optimized for combination studies, e.g., one may have one transgenic mouse model for dealing with senescent cell experiments, and another for epigenetic reprogramming experiments, and these may not be compatible or easy to fuse. (Using appropriate viral methods could overcome this but depends on good delivery vectors, and so forth.)
- This is not to mention the fact that combination studies will inherently require a larger number of subjects N to gather appropriate statistics, which as mentioned above seems to be hard for academic labs to achieve.
- Meanwhile, biotech and pharma are also not incentivized to do combinatorial studies.
- This is because pharma companies make money off of readily-translatable drugs, mostly small molecule drugs. Combinations would be harder to get approval for, especially if they involve new modalities like epigenetic reprogramming that may require more far-off inducible gene therapies, or unusual methods like blood dilution or combinations of multiple antibodies to block multiple age-associated targets. To make a simple and robust business case with a well-defined risk calculation, a pharma company wants simple single-target small molecule drugs, and that is simply not what the aging field, at its current level of development and possibly ever, requires to make progress.
- This leaves a gap where no organizations are seriously pursuing large-scale combination studies, to my knowledge, neither in humans or animals, and neither for advanced interventions like epigenetic reprogramming nor for simple lifestyle interventions, despite proposals.
- Biomarkers are not yet established to be causal, as opposed to correlative
- Epigenetic aging clocks and/or proteomic aging clocks would, in principle, show promise as primary endpoints for pre-clinical or clinical studies. Rather than waiting years to see extension of a mouse or human’s health-span, one could in a matter of weeks or months measure changes to epigenetic or proteomic clocks that are predictive of their ultimate healthspan.
- Yet the field of aging biomarkers still has major problems that limit this possibility at a technical level.
- Specifically, it is not yet known to what degree epigenetic or proteomic aging signatures are causal of aging, versus correlative. (There are some statistical reasons to worry they may not be causal, although probably this particular reasoning applies mostly to first gen studies that relied on patchy cross-sectional datasets like the Horvath multi-tissue clock; if one uses cohorts then one gets better predictors, which is part of why GrimAge and PhenoAge work so well for mortality.) This poses several problems:
- If they are only correlative, then there may be ways that putative therapies could “turn back the clocks”, but without affecting aging itself, i.e., they would only treat surface level indicators and thus be misleading
- In the worst case, the epigenetic and proteomic changes could represent compensations in the body acting against aging or to forestall aging. In that case, turning back the clocks might actually accelerate aging!
- Additionally, for epigenetic reprogramming therapies that probably operate at least in part through DNA methylation changes (and thus are visible in epigenetic clocks), the full set of damage types they reverse is not yet established. To my knowledge (and also Laura Deming’s), for example, nobody has measured whether the epigenetic reprogramming procedures used by Ocampo et al or the Sinclair lab will reduce the age-dependent buildup of lipofuscin aggregates in cells (whereas rapamycin seems to in some studies, as well as centrophenoxine). Likewise it would be nice to look at the shape/integrity of the nuclear lamina, and whether epigenetic changes repair this as well. There are probably many other examples of this sort.
- Finally, controversy about epigenetic clocks limits their adoption, so many studies coming out in different parts the aging field don’t measure this even though it would be easy to, e.g., the recent Conboy blood results don’t include an epigenetic clock measurement
- See here for a table of current putative limitations of epigenetic clocks.
- Aging itself is not yet established as a disease or as an endpoint for clinical trials, and this may exacerbate the systemic market failures that plague preventative medicine
- Since underlying aging factors likely cause many diseases in a multi-systemic fashion (e.g., systemic inflammation and circulatory damage may mediate both Alzheimer’s susceptibility and many other problems, e.g., cardiovascular, renal), and since these factors would best be dealt with preventatively, it would be ideal if aging itself could be classified as a disease, and even more ideal if changes in a predictive biomarker of aging could be used as an endpoint for trials
- Many aging researchers made this same point recently in an essay in Science.
- Yet the government is not moving on this issue, to my knowledge.
How to accelerate progress
I see four categories of systemic intervention here, all of which could be done under the umbrella of a coordinated ARPA-style initiative, at a scale of multiple tens of millions of dollars, that would cut across multiple sectors:
- Build and/or fund non-academic Focused Research Organizations, that would carry out large-N, combinatorial screens while assaying as many phenotypic features as possible.
- We can choose to view the problem from an engineering lens: as a “search” and “measurement” problem. Viewed from this lens, we should build a dedicated, appropriately-scaled, incentive-aligned, well-managed organization to carry out the required scalable assays and search procedures — or potentially create the impetus via milestone-based funding to pivot an existing organization to focus heavily on this (possibilities for existing organizations to re-focus on this could perhaps include aging gene therapy startups like Rejuvenate Bio, Gordian Biotechnologies, a biology experiment platform company like Strateos or Vium, or potentially an organization like the Buck Institute, could partially re-structure or expand to focus on this, or perhaps some partnership of such — there also appears to be at least one aging focused CRO).
- The purpose of such an organization could be: Searching the combinatorial space of aging interventions, while assaying the combinatorial space of aging phenotypes.
- While many individual aging mechanisms and their associated biology control knobs are being discovered in a piecemeal fashion, we do not yet have a way to comprehensively rejuvenate mammals or extend their lifespans. Achieving this likely requires not just turning one knob at a time but turning multiple independent and interacting knobs in the correct pattern. This leads to a large combinatorial search space, but to our knowledge no organization has yet tried to search it except on the smallest of scales, probably due to the mismatch of this problem with both short term corporate (low-risk single-drug-at-a-time development) and individual-academic-lab (competitive differentiation and credibility) incentives.
More on the combinatorial intervention aspect:
- Rather than immediately shooting for a single mechanism (what an academic would mostly be incentivized to do) or a single blockbuster drug molecule (what a company would mostly be incentivized to do), we need to first find some set of sufficient conditions for modulating healthspan by large “leaps” in the first place.
- We need a systematic project to apply factorial design to screen combination therapies across many mechanistic classes, in order to get a hit on at least one combined set of perturbations to mammalian organismal biology that can boost healthspan and regenerate many tissues without side effects.
- Even in a mouse, I think one could argue that this would revolutionize prospects in aging research and allow efforts to then be focused on more promising pathways — we would know at least one approach that works towards the end goal of broad healthspan extension, and the question would be translating it to a viable therapy, not whether it is fundamentally possible to slow, stop or reverse aging in a complex long-lived mammal.
- The “search process” could focus on simple endpoints like lifespan and serum-derived biomarkers, but alternatively, and preferably, it could comprehensively measure multi-system impacts (e.g., on all 9 “hallmarks of aging”) of interventions using multi-omics assays, and dynamically adapt the interventions accordingly, while building up a digital mapping between a multi-dimensional space of interventions and a multi-dimensional space of phenotypic effects.
- On the assay side, this could utilize, and in the process push forward, new technologies for scanning 3D biomolecular matter in a very general and precise way (e.g., combinations of FISSEQ, MERFISH, Expansion Microscopy, multiplexed antibody staining with DNA or isotope barcoded antibodies), where one can localize and identify many different molecules per specimen at high 3D spatial resolution and over large volumes of tissue, e.g., mapping the locations and identities of hundreds to thousands of disease-relevant proteins and RNAs throughout intact specimens at nanoscale resolution. The core chemistries and imaging technologies for this exist, but they need to be integrated and brought to scale.
- At this level of detail, we could ask questions like: How does the loss of synapse integrity in the aging brain, say, relate to the disruption of gene expression patterns in particular parts of the tissue, subcellular damage such as holes in the nuclear membranes inside cells or buildup of lipofuscin “junk”, changes to blood brain barrier integrity, or altered surveillance by the immune system? Rather than measuring these sparsely and separately, we could measure them comprehensively and integratively inside the same intact specimen.
- On the assay side, this could utilize, and in the process push forward, new technologies for scanning 3D biomolecular matter in a very general and precise way (e.g., combinations of FISSEQ, MERFISH, Expansion Microscopy, multiplexed antibody staining with DNA or isotope barcoded antibodies), where one can localize and identify many different molecules per specimen at high 3D spatial resolution and over large volumes of tissue, e.g., mapping the locations and identities of hundreds to thousands of disease-relevant proteins and RNAs throughout intact specimens at nanoscale resolution. The core chemistries and imaging technologies for this exist, but they need to be integrated and brought to scale.
- A more advanced version of the combinatorial intervention approach might use highly multiplexed CRISPR activation/inactivation of genes using libraries of CRISPR gRNAs and transgenic animals that already express the CRISPR machinery itself. Already, people are doing dozens of CRISPR sites per cell in other contexts, and reading out results based on whole-transcriptome RNAseq. This could be done first at a cellular level in a dish, and then at an organismal level using appropriate systemic gene delivery vectors, e.g., optimized blood brain barrier crossing AAVs or similar. See our review on in-vivo pooled screening for aging.
- Note that the company Rejuvenate Bio was founded on the basis of this PNAS paper on a combinatorial gene therapy approach in animals (3 non-cell-autonomous genes with individually known effects), but it appears that its go-to-market strategy is based on a fixed combination of just two of those gene therapy targets in dogs with a congenital cardiovascular disease. It is unclear to me if it is planning to do large scale screening of novel combinations or to target aging itself in the near term. Startups often need to take the shortest path to revenue.
- Outside of animal models, a program could also strongly pursue combinations of, e.g., already-FDA-approved compounds in human studies. Intervene Immune’s and Steve Horvath’s work with the TRIIM study is suggestive (but could be massively scaled, expanded and better outfitted), as is the TAME metformin trial. This would synergize nicely with a focused effort to improve epigenetic clock and other aging biomarkers, e.g., the Immune Risk Profile. Indeed, arguably an entire program could focus on human studies of systemic anti-aging / rejuvenation interventions that would fall short of a completely general approach to aging, e.g., combination therapies for immune system rejuvenation in the elderly.
- Fund an ARPA-style initiative to develop new tools for measurement and highly specific perturbation of aging phenotypes
- One notable low-hanging-fruit opportunity here is to lower the cost of genome-wide epigenetic clock measurements by 10x, while increasing their depth of mechanistic access
- Epigenetic clocks currently use methylation arrays, costing around $400 per sample. This limits the size of study that can be done, e.g., to do a 100k-1M people, or 1-10k people or animals with 30 tissue types per sample and 3 time points, would be cost-prohibitive here even for the largest organizations.
- The emergence of next-generation sequencing based methods for DNA methylation measurement (e.g., TAPS) would allow reduction to $10 per sample, via DNA-sequence-tag-based multiplexing of many samples into a single sequencing run, a 40x cost reduction
- Note that this could have major spinoff application to areas like liquid biopsy for cancer detection, some of which already rely on circulating tumor DNA (ctDNA) methylation, and wherein, because cancer incidence is low, very large N studies are needed to test for the statistical significance of an early detection method
- New epigenetic and proteomic profiling tools would also have major application to patient stratification or novel primary endpoints for clinical trials of diverse diseases
- One could also fund this to be applied at the single-cell level (definitely possible — see these papers) and in combination with recent methods for single-cell chromatin accessibility, single-cell RNAseq, single-cell chromatin conformation capture, or scRNAseq + CyTOF to measure both RNA and proteins at single-cell resolution. This would be likely to get to a far more mechanistic level of description than bulk methylation based epigenetic clocks. (An interesting form of clock indeed would be one that measured mismatches between DNA methylation, RNA and protein levels.)
- Next generation clocks based on proteomics, mtDNA, or exosomes could be powerful
- One could also invest in next generation proteomic profiling that goes beyond the SomaLogic targeted aptamer panels (on the order of a couple of thousand proteins assayed, out of 23k protein-coding genes in the genome not to mention many variants) to profile many more proteins or in a more unbiased way, and/or catalyze the use of such emerging technologies by the aging field by stimulating/funding collaborations with emerging companies in the proteomics field
- Mitochondrial DNA profiling could be relevant as well, e.g., single cell mitochondrial genome sequencing
- We could fund entirely new, and possibly even more powerful, methods of profiling aging, e.g., exosomal vesicles appear to provide a noninvasive transcriptome measure that could be applied in humans with a blood draw and methods are emerging for cell type specific exosome extraction, which could in theory perhaps get us a tissue-type or cell-type specific epigenetic clock using sampling of exosomal vesicles in circulation via sequencing.
- We could develop improved and more accessible ways to measure key aging-related metabolites like NAD (i.e., specific small molecules)
- We could stimulate the maturation and application to aging of in-situ spatially resolved epigenomics methods
- On the precise perturbation side, we could look into the state of the technology for multiplex targeted removal of specific factors from blood. This could also use a combinatorial approach, e.g., with combinations of inactivating aptamers or antibodies, either delivered to the animal or used as capture arrays for a blood filtration device. This could allow casual examination of the significance of potential detrimental factors found in aged blood plasma. We could also invest in other improved tools for tracking the causal influence of specific blood factors on aging in downstream tissues.
- We could also ensure that appropriate in-vivo multiplex CRISPR activation and inactivation technologies are available to meet the needs of the aging field.
- One notable low-hanging-fruit opportunity here is to lower the cost of genome-wide epigenetic clock measurements by 10x, while increasing their depth of mechanistic access
- Fund an ARPA-style initiative to create validated, causal epigenetic, proteomic, scRNAseq, exosomal or combined “clocks” and apply them to large existing banks of samples
- For epigenetic clocks, this could include:
- Single-cell studies to determine whether epigenetic clocks (in various tissues) reflect large changes to a sub-population of cells (e.g., adult stem cells), versus small changes to many types of cells indiscriminately, versus changes in the cell type composition of the tissue without changes to any individual cell type — this could help to establish the causal mechanisms, if any. From a recent paper: “DNA methylation-based age predictors are built with data from bulk tissues that represent a mix of different cell types. It is plausible that small changes in cell composition during ageing could affect the epigenetic age of a tissue. Stem cells, which generally decrease in number during ageing, might be one of these cell populations. Consequently, the epigenetic clock might be a measure of the different proportions of stem and differentiated cells in a tissue.” If true, this could provide an impetus to target epigenetic reprogramming efforts specifically toward ameliorating stem cell exhaustion.
- More generally, we can try to measure the mechanisms driving epigenetic clock “progression” over aging
- Decoupling damage from aging, e.g., understand what is unique about aging as opposed to other kinds of damage to cells, and that can be uniquely tracked by next-gen “clocks”. Cells can be exposed to various specific kinds of damage and repair to see the impact on the clocks, as opposed to the impact of aging as such, and clocks can be “focused” to specifically measure aging itself.
- Fund measurements determine whether epigenetic reprogramming therapies (e.g., pulsed OSK(M) as in Ocampo et al 2016) also restore other types of cellular damage beyond epigenetic states per se, e.g., buildup of lipofuscin, or decrease in proteo-stasis, and determine whether improved biomarkers can be devised that track all of these aspects as opposed to merely epigenetics
- Applying the resulting new, richer, cheaper biomarkers to existing bio-banks, including those that tracked people over long timescales and recorded their ages and causes of mortality, and releasing the resulting data in a machine-learning-accessible way such that anyone can train new predictive models on these data
- Existing cohorts (e.g., mentioned in Wyss-Coray papers) include the INTERVAL study, the SG90 Longevity Cohort study, the Einstein Aging Study cohort, Lothian Birth Cohorts (LBCs) of 1921 and 1936, the Whitehall study, Baltimore Longitudinal Study of Aging (BLSA), and probably many others.
- We would want to make all the data easily accessible in a universal database so lots of people with expertise in machine learning around the world could extract new kinds of predictors from it
- Developing proteomic or epigenetic profiles that are targeted to more specific known effects, like they recently did for senescence associated secretory phenotype (SASP) but much more broadly and aggressively. Other profiles could include a set of human homologues of Naked Mole Rat proteins thought to be involved in their aging resistance.
- For epigenetic clocks, this could include:
- Educate and incentivize the government to take appropriate actions
- Lobby the FDA to classify organismal senescence itself as a disease
- Perhaps this needs to be done by first educating members of Congress, Tom Kalil suggests
- Recent though partial progress has occurred with the TAME metformin trial, which had languished for years but recently appears to have accelerated due to a $40M infusion of private funding: “Instead of following a traditional structure given to FDA approved trials (that look for a single disease endpoint) TAME has a composite primary endpoint – of stroke, heart failure, dementia, myocardial infarction, cancer, and death. Rather than attempting to cure one endpoint, it will look to delay the onset of any endpoint, extending the years in which subjects remain in good health – their healthspan.” It may be possible to replicate this trial design for future studies.
- Push for more aging-related biomarker endpoints in clinical trials of all sorts of drugs
- Lobby for expansion of the NIH Interventions Testing Program (ITP) for aging drugs, or perhaps for NIA to fund external such programs — including going to more complex treatments beyond small molecules, and replicating key aging studies at large N
- Create funding-tied incentives for larger-N and replicated studies in the aging field, as well as for certain phenotypic measurements to become common and released openly for all funded studies
- Lobby the FDA to classify organismal senescence itself as a disease
Acknowledgements:
Thanks to Sarah Constantin, Laura Deming and Sam Rodriques for helpful discussions prior to the writing of this document.